Miyelodisplastik Sendrom (MDS) – Kısa Bilgiler
MDS olarak da adlandırılan miyelodisplastik sendrom, hematopoetik sistemin (kan yapıcı sistem) bir hastalığıdır. İleri aşamalarda lösemi gelişimi mümkündür. Aşağıdaki yazıda hastalık tablosu, olası hastalık sebepleri ve bulguları, hastalığın seyri, tanısı, tedavi planlaması, tedavisi, tedavi iyileştirme çalışmaları, tedavi sonrası izlemi ve hastalığın prognozu hakkında detaylı bilgi bulacaksınız.
yazar: PD Dr, med Ayami Yoshimi, editör: Maria Yiallouros, Yayın İzni: Prof. Dr. med. Charlotte Niemeyer, Dr. Miriam Erlacher, türk tercüman: Dr. med. Ebru Saribeyoglu, Last modification: 2024/04/17 https://kinderkrebsinfo.de/doi/e221512
Table of contents
Hastalık tablosu
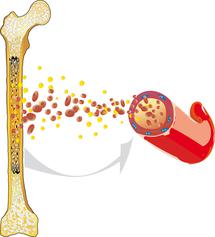
Miyelodisplastik sendrom (kısaca MDS), kemik iliğinin normal çalışmadığı ve bu nedenle yeterli sayıda sağlıklı kan hücresinin üretilmediği bir grup hastalıktır.
Kırmızı kan hücreleri (eritrositler), beyaz kan hücreleri (lökositler) ve trombositler (kan pulcukları) dahil olmak üzere kanda bulunan tüm kan hücreleri, kan kök hücreleri olarak adlandırılan kemik iliğindeki kan yapıcı hücrelerden kaynaklanır. Kemik iliğindeki kan kök hücrelerinden işlevsel kan hücrelerinin oluşabilmesi için çok sayıda olgunlaşma ve bölünme süreci gerekir. Uzmanlar bu süreçlere hematopoez adını vermektedirler. Hematopoez hakkında daha fazla bilgiyi kemik iliği ve kanın yapısı ve işlevi hakkındaki hasta bilgilendirme yazımızda bulabilirsiniz.
MDS'li hastalarda kemik iliğindeki bu süreçler bozulur. Hücreler hatalı olgunlaşır ve mikroskop altında sağlıklı hücrelerden farklı görünür (displazi). Çoğu zaman bu hatalı şekillenmiş hücreler kemik iliğinde ölür, böylece buradan kana yeterince kan hücresi aktarılamaz. Sonuç olarak, kanda yeterince sağlıklı kırmızı ve beyaz kan hücresi ve trombosit bulunmaz ve bu da kansızlık (anemi), enfeksiyonlar veya kanama eğiliminin artması gibi çeşitli sağlık sorunlarına neden olur.
Öte yandan, kan kök hücrelerinin olgunlaşmasının bozulması, kemik iliğinde olgunlaşmamış kan hücrelerinin (blast olarak da adlandırılırlar) sürekli olarak artmasına neden olur. Olgunlaşmamış olmaları nedeniyle bu hücreler beklenen işlevlerini yerine getiremezler. Bazı MDS hastalarında kemik iliği ve kandaki blast [blastlar] sayısı hastalığın seyri sırasında o kadar artar ki MDS artık lösemiden ayırt edilemez hale gelir. Bu nedenle MDS eskiden "prelösemi" (lösemi öncüsü) olarak adlandırılırdı.
Görülme sıklığı
Miyelodisplastik sendrom (MDS) yaşlılarda kemik iliğinin en sık görülen malign hastalığıdır; çocuklarda ve ergenlerde çok nadir görülür. Çocuklarda ve 18 yaş altı ergenlerde görülen tüm kan kanserlerinin yaklaşık %8'ini ve bu yaş grubundaki tüm kötü huylu hastalıkların yaklaşık %2,5'ini oluşturur.
Alman Çocukluk Çağı Kanser Kayıt Merkezine (Mainz) göre, Almanya'da her yıl 0-17 yaş arası yaklaşık 54 hastaya yeni MDS tanısı konulmaktadır. Ortalama görülme yaşı yaklaşık 9'dur. Erkekler kızlardan biraz daha sık etkilenmektedir (cinsiyet oranı: 1,2 : 1).
Miyelodisplastik sendrom çeşitleri
Miyelodisplastik sendromun (MDS) gelişim şekline bağlı olarak ve kan ve kemik iliğinin mikroskobik incelemeleri ışığında, aşağıda belirtilen farklı MDS çeşitleri bulunmaktadır.
Primer (birincil) ve sekonder (ikincil) MDS
Miyelodisplastik sendrom (MDS) genellikle görünürde bir neden olmaksızın ortaya çıkar. Buna "birincil" MDS denir. MDS'li çocukların yaklaşık %75'i bu gruba dahildir. Bununla birlikte, çocukluk çağındaki "birincil" MDS'de henüz tanımlanmamış olan doğuştan genetik değişikliklerin de mevcut olabileceği varsayılmaktadır (ayrıca bkz. "Sebepleri" bölümü).
Bazı hastalarda MDS gelişimi belirli tetikleyicilerle bağlantılı olabilir; bu durumlarda "ikincil" MDS'den söz edilir. Örneğin, bazı hastalarda hastalık, genellikle kötü huylu başka bir hastalık nedeniyle uygulanan radyoterapi (ışın tedavisi) veya kemoterapi tedavisinden sonra gelişir. Diğer hastalar, MDS tanısı konmadan önce kemik iliği fonksiyon bozukluğuyla ilişkili doğuştan bir hastalığa sahiptir. Bunlar arasında Fanconi anemisi, diskeratozis konjenita, Shwachman-Diamond sendromu, Diamond Blackfan anemisi (DBA) veya ağır konjenital nötropeni sayılabilir. Edinilmiş aplastik anemi (SAA) de MDS tanısından önce ortaya çıkabilir.
DSÖ sınıflandırmasına göre primer çocukluk çağı MDS alt tipleri
Çocuklarda birincil MDS, Dünya Sağlık Örgütü'nün (DSÖ; ingilizcesi World Health Organization, WHO) sınıflandırmasına göre iki alt tipe ayrılır DSÖ sınıflaması. Temel olarak kan ve kemik iliğindeki olgunlaşmamış hücrelerin (blastlar) oranını referans alır.
Çocukluk çağı dirençli sitopenisi (RCC)
"Çocukluk çağının dirençli sitopenisi" (RCC) olarak adlandırılan hastalarda, kanda %2'den az ve kemik iliğinde %5'ten az olan blast [blastlar] yüzdesi sağlıklı çocuklara kıyasla önemli ölçüde artmamıştır. Bununla birlikte, sıklıkla, RCC'li çocukların kemik iliğinde hücre sayısı oldukça düşüktür, yani normalde kemik iliğinde bulunan tüm hücreler (kırmızı kan hücreleri, beyaz kan hücreleri, trombositler) azalmış sayıda bulunur. Bu nedenle, ayırıcı tanı olarak adlandırılan ayırıcı tanının bir parçası olarak, kemik iliğinin başka bir konjenital hastalığının (kemik iliği yetmezliği) veya aplastik aneminin mevcut olup olmadığı her zaman açıklığa kavuşturulmalıdır, çünkü bu hastalıklarda da hücre bakımından yetersiz bir kemik iliği söz konusudur.
Blast fazlalığı ile seyreden miyelodisplastik sendrom (MDS-EB)
MDS hastalarında kan ve kemik iliğindeki blast [blastlar] oranı sırasıyla %2 ve %5'in çok üzerine çıkabilir. Bu durum MDS-EB, yani aşırı blastlı MDS (EB) olarak adlandırılır. Ancak kemik iliğindeki blast oranı %29'u geçmemelidir, çünkü bu durumda lösemi söz konusudur. Ancak MDS ile lösemi arasında ayrım yapılırken, örneğin blast sayısının artış hızı gibi başka faktörler de dikkate alındığından, MDS-EB ile lösemi arasında ayrım yapmak (ayırıcı tanı) bazen zor olabilmektedir. Sınırdaki vakalarda, yani blast sayısının %20 ila 30 arasında olduğu durumlarda, çeşitli tanı yöntemleriyle tanıyı doğrulamak için iki hafta sonra kemik iliği ponksiyonunu tekrarlamak gerekir.
|
MDS alt tipi |
Kan |
Kemik iliği |
|---|---|---|
|
Çocukluk çağının dirençli sitopenisi (RCC) |
Blastlar dahar az %2 |
Blastlar dahar az %5 |
|
Blast fazlalığı ile seyreden miyelodisplastik sendrom (MDS-EB) |
Blastlar %2 – 29 |
Blastlar %5 – 29 |
Sebepleri
Miyelodisplastik sendromun (MDS) kesin nedenleri genellikle açıklanamamıştır; ancak hastalık – diğer kanser türleri gibi – ne bulaşıcıdır ne de diğer insanlara geçirilebilir. Çoğu vakada, MDS önceden sağlıklı olan çocuklarda görünürde bir neden olmaksızın gelişir ("birincil MDS" olarak adlandırılır). Bununla birlikte, genellikle bu hastalığa yakalananların MDS veya lösemi gelişimine karşı özel bir duyarlılığa (yatkınlık) sahip olduğu varsayılır. Bu yatkınlık "predispozisyon" olarak da adlandırılır.
Yatkınlık, sperm veya yumurta hücrelerinde (yani germline/germ, üreme/eşey hattında) de bulunan ve bu nedenle kalıtsal olabilen gen değişikliklerinden (mutasyon) kaynaklanır. Bu durumda, gen değişikliği hastanın tüm vücut hücrelerinde de mevcuttur. Etkilenen çocuklar ya bu germ hattı (eşey, üreme hattı) mutasyonunu bir veya her iki ebeveynden miras almışlardır ya da hastalıklı kişinin gen değişikliği döllenmiş yumurta hücresinde yeni gelişmiştir.
Kemik iliğinde doğuştan hematopoez bozukluklarına neden olabilen gen mutasyonlarının (örneğin Fanconi anemisi, ağır konjenital nötropeni, diskeratozis konjenita veya Diamond Blackfan anemisi) MDS oluşumuna yatkınlıkla da ilişkili olduğu uzun yıllardır bilinmektedir. Son yıllarda burada rol oynayan başka kalıtsal hastalıklar da tespit edilmiştir. Bunlar arasında GATA2 eksikliği veya SAMD9/SAMD9L sendromu sayılabilir. Bu doğuştan gelen hastalıklar genellikle artmış kanser riski ile ilişkili olduğundan, kansere yatkınlık yaratan sendromlar olarak da adlandırılırlar.
Yaşlı insanlarda MDS'nin gelişimi farklıdır. Çoğu yetişkin vakada doğuştan gelen bir sorun yoktur, bunun yerine on yıllar boyunca edinilen gen değişiklikleri bir hücreyi yavaş yavaş MDS veya lösemi hücresine dönüştürür. Bazı hastalar MDS tanısı konmadan önce başka bir kanserin tedavisinin bir parçası olarak radyoterapi veya kemoterapi almışlardır. Bu durumda MDS, en azından kısmen ilk kanserin tedavisi tarafından tetiklenen ikinci hastalıktır (ikincil MDS).
Hastalık belirtileri
Miyelodisplastik sendromlu (MDS) bir hastada ortaya çıkabilecek belirtiler (semptomlar) öncelikle (işlevsel) kan hücrelerinin eksikliğinin ne kadar ağır olduğuna, yani sitopeninin derecesine bağlıdır. Kanın hangi hücrelerinin etkilenmiş olduğuna bağlı olarak, aşağıdaki sitopeni biçimleri ve bununla ilişkili olarak aşağıdaki semptomlar tanımlanabilir:
Kansızlık: Kırmızı kan hücrelerinin eksikliği (anemi)
Kırmızı kan hücrelerinin (eritrositler) görevi, nefes alırken akciğerler yoluyla alınan oksijeni vücudun çeşitli organ ve dokularına taşımaktır. Kırmızı kan hücrelerinin eksikliği (anemi) solgunluk, yorgunluk, halsizlik ve baş ağrısı gibi şikayetlere yol açar.
Bağışıklık sisteminin zayıflığı: Beyaz kan hücrelerinin eksikliği (lökopeni/nötropeni)
Beyaz kan hücreleri (lökositler) patojenlere karşı savunmadan ve dolayısıyla enfeksiyonların önlenmesinden sorumludur. Bağışıklık sisteminin savunma mekanizmasında farklı görevleri yerine getiren lenfosit lökositler [lenfositler] ve granülosit [granulositler] gibi farklı lökosit çeşitleri vardır. Lökopeni [lökopeni] olarak adlandırılan işlevsel beyaz kan hücrelerinin eksikliği nedeniyle vücut enfeksiyon riski altındadır. MDS'li hastalarda granülosit sayısı genellikle özellikle azalır (granülositopeni veya nötropeni olarak adlandırılır). Bunlar bakteri ve mantarlara karşı savunmadan sorumlu olduklarından, MDS hastalarında özellikle ateş şeklinde kendini belli eden bakteri ve mantar enfeksiyonları ortaya çıkar.
Kanamaya yatkınlık: Kan pulcuklarında azalma (trombositopeni)
Kan pulcukları (trombositler) kanın pıhtılaşmasında [kan pıhtılaşması] önemli bir rol oynarlar. Trombositler azalırsa (trombositopeni olarak adlandırılır), hem kendiliğinden hem de yaralanmalardan sonra kanama daha hızlı gerçekleşir. Bu, örneğin deride veya mukozada noktasal kanamalar (peteşi), çürükler (hematom) ve/veya burun veya diş kanaması şeklinde kendini gösterir, ancak iç organlarda veya beyinde ciddi kanamalar da meydana gelebilir. Trombositopeni ne kadar derinse, yani trombosit eksikliği ne kadar fazlaysa, o kadar yüksektir.
Blast fazlalığı olan MDS (MDS-EB) (ayrıca bkz. Bölüm "Miyelodisplastik sendrom çeşitleri") hastalarında, küçük kan damarlarının iltihaplanması veya ateş ve ciltte kırmızı yumrularla seyreden Sweet sendromu gibi tedavisi zor belirtiler görülebilir.
Tanı
Doktor veya çocuk doktoru muayene ettiği çocuğun hastalık geçmişinde (anamnezi) ve fiziksel muayene kapsamında bir kan hastalığı söz konusu olabileceğine dair veriler elde ederse, önce geniş kapsamlı bir kan tetkiki yaptıracaktır. Kan sayımı [kan tablosu] içindeki bazı değişiklikler miyelodisplastik sendrom gibi bir kan hastalığı şüphesini arttırırsa, teşhisin yani tanının güvencesi açısından kemik iliği (kemik iliği ponksiyonu, kemik iliği aspirasyonu) örneği alınması gerekli olacaktır. Bu amaçla ve daha sonra yapılacak olası tetkikler için doktor, hastasını çocuklar ve gençlerde kanser ve kan hastalıkları alanında uzmanlaşmış bir hastaneye sevk edecektir (pediatrik onkoloji ve hematoloji kliniği).
Kan ve kemik iliği muayenesi
MDS hastalarında görülen belirtilerin kesin olarak MDS'ye işaret etmemesi ve lösemiler gibi diğer kan hastalıklarında da ortaya çıkması nedeniyle, bu hastalığın tanısı ancak kan ve kemik iliğinin kapsamlı bir şekilde incelenmesiyle konulabilir.
MDS'nin kesin tanısı için leğen (pelvis) kemiğinden alınan kemik iliğine ihtiyaç vardır. Bunun için kemik iliği aspirasyonu (kemik iliği ponksiyonu) ve kemik iliği biyopsisi yapılması gerekir. Her iki yöntem de birbirini tamamlar ve ortak bir tetkiğin parçası olarak gerçekleştirilir. Kemik iliği aspirasyonunda (ponksiyonunda) kemik özel bir içi boş iğne ile delinir ve az miktarda kemik iliği kanı çekilir. Kemik iliği biyopsisinde ise içi boş bir iğne ile leğen (pelvis) kemiğinden küçük bir parça (yaklaşık 1 mm çapında) alınır. Kemik iliği ponksiyonu ve kemik iliği biyopsisi hakkında daha fazla genel bilgiyi burada bulabilirsiniz.
Örnek alındıktan sonra kemik iliğindeki kan yapıcı hücrelerin görünümü, kan ve kanser hastalıkları uzmanı tarafından mikroskop altında değerlendirilir. MDS durumunda, hücrelerin görünümünde tanı için belirleyici olan çeşitli değişiklikler (displazi) görülebilir. Blast fazlalığı olan miyelodisplastik sendromda (MDS-EB), kemik iliğindeki blast hücrelerinin (blastlar] sayısı artmıştır (bkz. Bölüm "Miyelodisplastik sendrom çeşitleri"). Kemik iliği aspiratından (ponksiyonundan) alınan materyal üzerinde kromozom incelemesi (sitogenetik) yapılır (aşağıya bakınız). MDS'dan şüpheleniliyorsa, güvenilir bir tanı için kemik iliği aspirasyonu (ponksiyonu) ve kemik iliği biyopsisi yaklaşık 14 gün sonra tekrarlanmalıdır.
Kromozomların incelenmesi (sitogenetik)
Blast fazlalığı olan MDS'li (MDS-EB) çocukların yarısından fazlasında ve refrakter sitopenili (RCC) hastaların yaklaşık %30'unda, kemik iliği ve kan hücrelerinde kromozom değişiklikleri saptanır. Bu değişikliklerin saptanması MDS tanısının doğrulanmasına yardımcı olur. MDS'li çocuklarda en yaygın ve tipik kromozomal değişiklik kromozom 7 kaybıdır (monozomi 7 olarak bilinir). Bu olgularda, kromozom 7 hücrelerde iki kez yerine yalnızca bir kez bulunur.
Moleküler genetik incelemeler
MDS'li bazı çocuk ve gençlerde hastalığa karşı genetik bir yatkınlık (predispozisyon), yani bir eşey hücresi (germ hücresi, üreme hücresi, germline) mutasyonu vardır (bkz. Bölüm "Sebepleri"). Bu genetik değişikliklerin belirlenmesi için moleküler genetik yöntemlerle kan ve/veya kemik iliğine ek olarak saç kökü hücreleri gibi diğer vücut dokularının da eşey hücrelerinin incelenmesi gerekir. MDS'li bir hastada bir eşey hücresi mutasyonu (germ mutasyonu) tespit edilirse, kök hücre nakli (KHN) planlanlandığı zaman, nakil için söz konusu olan aile içi vericide de bu mutasyon aranmalıdır. Ancak aile içi verici adayında bu mutasyon tespit edilemezse bu kişi kök hücre bağışında bulunabilir.
Tedavi
Miyelodisplastik sendromlu (MDS) hastaların tedavisi hastalığın şekline, yani birincil (primer) veya ikincil (sekonder) MDS olup olmadığına veya primer MDS durumunda çocukluk çağının refrakter sitopenisi (RCC) veya blast fazlalığı olan MDS (MDS-EB) olup olmadığına bağlıdır (bkz. Bölüm "Miyelodisplastik sendrom çeşitleri"). Genel olarak tedavi için aşağıdaki seçenekler mevcuttur:
- yüksek doz kemoterapi sonrası (kondisyon tedavisi / hazırlama tedavisi) bir vericiden alınan hematopoetik kan hücrelerinin nakli (allojenik kemik iliği nakli, KHN veya KİN)
- immunsupresif tedavi (bağışıklık sistemini baskılayıcı tedavi, IST)
- diğer ilaç tedavileri: kemoterapi, Azasitidin
- destek tedavisi: transfüzyon (kan nakli), antibiyotik
Aşağıda, farklı MDS formlarına yakalanmış hastaların tedavisi anlatılmaktadır.
Refrakter çocukluk çağının dirençli sitopenisi (RCC)
Refrakter çocukluk çağı sitopenisi (RCC) olan hastalar, kan ve kemik iliği hücrelerinde kromozomal değişikliklerin varlığına veya yokluğuna, kandaki hücre azalmasının derecesine (sitopeni) ve kemik iliğinin hücre içeriğine (hücresellik) bağlı olarak farklı şekilde tedavi edilir.
Kemik iliği hücrelerinin kromozom analizinde kromozom 7 kaybı (monozomi 7) saptanan RCC hastalarının bir ila iki yıl içinde blast hücresi proliferasyonu riski ile birlikte kötü bir hastalık seyri göstermesi beklenir. Bu nedenle bu hastalara mümkün olan en kısa sürede allojenik kemik iliği nakli (KİN veya KHN) yapılmalıdır. Bu tedavide, önce kemik iliğindeki tüm hücreleri (sağlıklı ve hastalıklı) yok etmek, başka bir deyişle kemik iliğini boşaltmak için hastaya hazırlama (kondisyon tedavisi, yüksek doz kemoterapi) adı verilen yüksek doz sitostatik ilaçlar verilir. Daha sonra, bir vericinin kemik iliğinden veya periferik kanından alınan sağlıklı kan kök hücreleri alıcıya nakledilir.
Buna karşılık, kromozomal bulguları normal olan RCC'de klinik tablo yıllarca değişmeden kalabilir. Bu hastalarda kan nakli gerekmiyorsa ve yeterli granülosit (bakteri ve mantarlarla savaşan beyaz kan hücreleri) varsa, başlangıçta tedavi edilmeden, sadece gözlemlenmekte ve düzenli (kan) tetkikleri yoluyla hastalığın seyri izlenmektedir ("izle ve bekle" yöntemi olarak adlandırılmaktadır). Ancak kan değerleri zamanla kötüleşirse, hastalığın seyri sırasında bir tedavi, genellikle de kök hücre nakli gerekli olabilir.
Tanı konulduktan sonra kan nakline ihtiyaç duyan veya kanında çok az granülosit bulunan normal kromozomal bulgulara sahip hastalar mümkün olan en kısa sürede kök hücre nakli ile tedavi edilir. Sağlıklı kardeşler veya uygun yabancı bireyler kök hücre vericisi olarak kabul edilebilir. Uygun kardeş verici bulunamazsa, hiposelüler (az hücreli) kemik iliği olan hastalar için immunsupresif tedavi (bağışıklık sistemini baskılayıcı tedavi, IST) tercih edilebilir. Bu immunsupresif tedavi, şiddetli aplastik anemi (SAA) [tedavisine benzer ve birkaç ay veya yıl sürer].
İmmunsupresif tedavi (IST) ile hastaların yaklaşık yarısında kan hücresi sayısında yeterli bir artış sağlanabilir, böylece kan transfüzyonu ihtiyacı ortadan kalkar. Başlangıçta başarılı olan IST'den sonra sitopenin-yani kan hücresi eksikliğinin tekrar gelişmesi de mümkündür (nüks/rezidiv) olarak da adlandırılır). Nüks veya IST tedavisine yanıt alınamaması durumunda, tam uyumlu yabancı bir vericiden kök hücre nakli önerilir.
MDS blast artışı ile seyreden MDS (MDS-EB)
Blast fazlalığı olan MDS hastalarında (MDS-EB), yani hastalığın ilerlemiş formunda, hastalığın ilerleyen dönemlerinde lösemi gelişme riski çok yüksektir. Bir MDS-EB için erken kök hücre nakli (kemik iliği nakli) genellikle tercih edilen tedavidir. Sağlıklı kardeşler veya tam uygun akraba olmayan bireyler kök hücre vericisi olarak düşünülebilir. Yüksek blast sayıları varlığında, nakil öncesi yüksek doz tedavi (kondisyon tedavisi) ve blastları azaltmak için kemoterapi ile ön tedavi yararlı olabilir.
İkincil (sekonder) MDS
Sekonder (ikincil) miyelodisplastik sendromlu (MDS) hastalar genellikle blast fazlalığı olan birincil MDS'lu (MDS-EB) hastalar gibi tedavi edilir (bkz. önceki bölüm). Bu nedenle bu hastalara tanıdan sonra mümkün olan en kısa sürede uygun bir kök hücre vericisinden nakil yapılmalıdır.
Tedavi sırasında destek tedavisi (suportif tedavi)
Miyelodisplastik sendromlu (MDS) tüm hastalar için destekleyici tedaviler (destek tedavisi) yararlı ve gereklidir. Bu ek uygulamalar hastalıkla ilişkili belirtilerin ve tedaviyle ilişkili yan etkilerin tedavisinde veya önlenmesinde yardımcı olur.
Hastaların çoğunda tanı anında anemi ve/veya trombosit eksikliği (trombositopeni) vardır ve bunlar çeşitli sağlık sorunlarına yol açabilir (bkz. Bölüm "Hastalık belirtileri"). Bu problemler kan nakli (kırmızı kan hücresi veya trombosit verilmesi) ile tedavi edilebilir. Bununla birlikte, tekrarlanan kırmızı kan hücresi nakli vücuda büyük miktarda demir yükü oluşturur, bu da zamanla organlarda (özellikle karaciğer ve kalp) birikerek hasara yol açabilir (aşırı demir yüklenmesi olarak da adlandırılır). Bu nedenle aşırı demir yüklemesi olan hastalar demirden yoksun bırakma tedavisi görmelidir.
MDS'nin kendisi ve aynı zamanda tedavisi nedeniyle, örneğin kök hücre nakli veya immunsupresif (bağışıklık sistemini baskılayıcı) tedavi, hastaların savunma mekanizmasını zayıflatır. Bu nedenle hastalar enfeksiyonlardan mümkün olan en iyi şekilde korunmalı ve bir enfeksiyon durumunda mümkün olan en hızlı şekilde tedavi edilmelidirler. Bağışıklık sistemi baskılanmış çocuklarda enfeksiyonlar her zaman hayati tehlike oluşturacak nitelikte kabul edilmelidir.
Bir enfeksiyonun ilk belirtisi genellikle ateştir. Bu nedenle ateşi yükselen hasta
ların ebeveynleri veya yakınları, çocukların derhal geniş kapsamlı antibiyotiklerle tedavi edilebilmesi için derhal (geceleri de) ilgili kliniğe veya doktora başvur-malıdır. Granülosit eksikliği (nötropenisi) olan hastalarda, destek tedavisi olarak koruyucu antibiyotik ve antifungallerin (antimikotikler) uygulanması gereklidir.
Tedavi başarısı
Miyelodisblastik sendromun (MDS) seyri büyük farklılıklar gösterebilir. Uzun bir süre boyunca dengede kalan hastalıklar vardır, bazıları ise hızla ilerler. Hastanın tahmini seyri (prognozu) hastalığın tipine (alt tipine) (bkz. Bölüm "Miyelodisplastik sendrom çeşitleri") ve kemik iliği hücrelerinde olası genetik değişikliklerin varlığına göre belirlenir.
Çocukluk çağının dirençli sitopenisi (RCC)
Çocukluk çağının dirençli sitopenisi (RCC) olan hastalar, belirli risk faktörlerinin varlığına veya yokluğuna bağlı olarak farklı tedavi gruplarına ayrılır ve böylece farklı tedaviler alırlar (gözlem, kök hücre nakli veya immunsupresif tedavi, bkz. Bölüm "Tedavi"). Bu hastalar, tüm tedavi gruplarında, %80-90 sağkalım olasılığı ile genel olarak iyi bir seyre (prognoza) sahiptir. Bu durum, monozomi 7 nedeniyle ilerlemiş MDS veya erken dönemde lösemiye dönüşme riski yüksekliği nedeni ile allojenik kök hücre nakli yapılan hastalarda için de geçerlidir Böyle bir tedavi ile prognoz diğer RCC hastaları ile aynı derecede iyidir.
RCC durumunda, kök hücre naklinden sonra hastalığın tekrarlama (nüks, rezidiv) riski çok düşüktür. Ancak her kök hücre nakli, hastalıklı kemik iliğinin yüksek doz kemoterapi sonrasında bir vericinin sağlıklı kemik iliği ile değiştirildiği yoğun bir tedavidir. Bu nedenle hastalar, kök hücre naklinden sonraki yaşamları boyunca, uzun dönem yan etkilerin zamanında fark edilip tedavi edilebilmesi için yılda en az bir kez kontrol muayenelerinden geçmelidir.
İmmunsupresif tedavi (IST, bağışıklık sistemini baskılayıcı tedavi) ile başarılı bir şekilde tedavi edilen veya tedavisiz gözlenen hastalarda bile, hastalığın nüksetmesini veya ilerlemesini erken bir aşamada saptayabilmek için kan sayımının düzenli olarak kontrol edilmesi ve kemik iliğinin, kemik iliği biyopsisi ve sitogenetik yöntemlerle yıllık olarak incelenmesi gerekir.
İlerlemiş MDS (MDS-EB) veya ikincil (sekonder) MDS
Blast fazlalığı olan MDS (MDS-EB) veya ikincil MDS'a sahip olan çocuklar ve gençler için allojenik kemik iliği nakli tek küratif (iyileştirici) tedavi seçeneğidir. Hastaların yaklaşık %50-60'ı bu tedavi ile iyileştirilebilir. Ciddi kromozomal değişikliği olan hastalar, yani üç veya daha fazla kromozomal değişiklik (kompleks karyotip olarak adlandırılır), oldukça olumsuz bir seyre (prognoza) sahiptir.
 Miyelodisplastik Sendrom (MDS) - Kısa Bilgiler (PDF) (311KB)
Miyelodisplastik Sendrom (MDS) - Kısa Bilgiler (PDF) (311KB)Kaynakça 
- Rudelius M, Weinberg OK, Niemeyer CM, Shimamura A, Calvo KR: The International Consensus Classification (ICC) of hematologic neoplasms with germline predisposition, pediatric myelodysplastic syndrome, and juvenile myelomonocytic leukemia. Virchows Archiv : an international journal of pathology 2023, 482: 113 [PMID: 36445482]
- Bortnick R, Wlodarski M, de Haas V, De Moerloose B, Dworzak M, Hasle H, Masetti R, Starý J, Turkiewicz D, Ussowicz M, Kozyra E, Albert M, Bader P, Bordon V, Cario G, Beier R, Schulte J, Bresters D, Müller I, Pichler H, Sedlacek P, Sauer MG, Zecca M, Göhring G, Yoshimi A, Noellke P, Erlacher M, Locatelli F, Niemeyer CM, Strahm B, for EWOG-MDS: Hematopoietic stem cell transplantation in children and adolescents with GATA2-related myelodysplastic syndrome. Bone marrow transplantation 2021, E-pub ahead of printg [PMID: 34244664]
- Erdmann F, Kaatsch P, Grabow D, Spix C: German Childhood Cancer Registry - Annual Report 2019 (1980-2018). Institute of Medical Biostatistics, Epidemiology and Informatics (IMBEI) at the University Medical Center of the Johannes Gutenberg University Mainz 2020 [URI: https://www.kinderkrebsregister.de/ typo3temp/ secure_downloads/ 42507/ 0/ 1c5976c2ab8af5b6b388149df7182582a4cd6a39/ Buch_DKKR_Jahresbericht_2019_komplett.pdf]
- Niemeyer C, Kratz C: Myelodysplastische Syndrome. in Niemeyer C, Eggert A (Hrsg): Pädiatrische Onkologie und Hämatologie 2018, Springer Verlag; 715 [ISBN: 978-3-662-43685-1]
- Niemeyer C, Eggert A (Hrsg): Pädiatrische Hämatologie und Onkologie. Springer-Verlag GmbH Deutschland 2. vollständig überarbeitete Auflage 2018 [ISBN: 978-3-662-43685-1]
- Ripperger T, Bielack SS, Borkhardt A, Brecht IB, Burkhardt B, Calaminus G, Debatin KM, Deubzer H, Dirksen U, Eckert C, Eggert A, Erlacher M, Fleischhack G, Frühwald MC, Gnekow A, Goehring G, Graf N, Hanenberg H, Hauer J, Hero B, Hettmer S, von Hoff K, Horstmann M, Hoyer J, Illig T, Kaatsch P, Kappler R, Kerl K, Klingebiel T, Kontny U, Kordes U, Körholz D, Koscielniak E, Kramm CM, Kuhlen M, Kulozik AE, Lamottke B, Leuschner I, Lohmann DR, Meinhardt A, Metzler M, Meyer LH, Moser O, Nathrath M, Niemeyer CM, Nustede R, Pajtler KW, Paret C, Rasche M, Reinhardt D, Rieß O, Russo A, Rutkowski S, Schlegelberger B, Schneider D, Schneppenheim R, Schrappe M, Schroeder C, von Schweinitz D, Simon T, Sparber-Sauer M, Spix C, Stanulla M, Steinemann D, Strahm B, Temming P, Thomay K, von Bueren AO, Vorwerk P, Witt O, Wlodarski M, Wössmann W, Zenker M, Zimmermann S, Pfister SM, Kratz CP: Childhood cancer predisposition syndromes-A concise review and recommendations by the Cancer Predisposition Working Group of the Society for Pediatric Oncology and Hematology. American journal of medical genetics. Part A 2017, 173: 1017 [PMID: 28168833]
- Yoshimi A, Strahm B, Baumann I, Furlan I, Schwarz S, Teigler-Schlegel A, Walther JU, Schlegelberger B, Göhring G, Nöllke P, Führer M, Niemeyer CM: Hematopoietic stem cell transplantation in children and young adults with secondary myelodysplastic syndrome and acute myelogenous leukemia after aplastic anemia. Biology of blood and marrow transplantation 2014, 20: 425 [PMID: 24316460]
- Niemeyer C: Myelodysplastic syndrome and aplastic anemia in children. Europ School of Oncology 2000, 31