Myelodysplastic Syndrome (MDS) – Brief information
Myelodysplastic syndromes (MDS) are diseases of the blood-forming system in the bone marrow. Advanced types of MDS may result in leukaemia. This text provides information about the characteristics and subtypes of the disease, its frequency, causes, symptoms, diagnoses, treatment, and prognosis.
Author: PD Dr, med Ayami Yoshimi, Editor: Maria Yiallouros, Reviewer: Prof. Dr. med. Charlotte Niemeyer, Dr. Miriam Erlacher, English Translation: Dr. med. Gesche Riabowol (nee Tallen), Last modification: 2024/05/30 https://kinderkrebsinfo.de/doi/e221512
Table of contents
General information on the disease
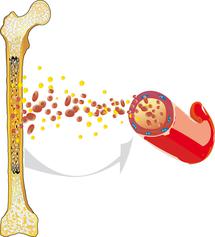
Myelodysplatic syndrome (briefly MDS) involves a group of diseases with impaired bone marrow function and subsequently insufficient production of red blood cells.
All blood cells in the blood – these include red blood cells (erythrocytes), white blood cells (leukocytes) and platelets (thrombocytes) – arise from the blood forming cells in the bone marrow, the so-called blood stem cells. Before the blood stem cells in the bone marrow become functioning blood cells, they must undergo multiple differentiating and dividing processes. Specialists call this haematopoiesis.
In patients with MDS, these processes in the bone marrow are impaired. The cells maturate defectively, they also look different under the microscope when compared to healthy cells (dysplasia). These defective cells often die already in the bone marrow, and thus, not enough blood cells enter the blood stream. As a consequence, there are not enough red and white blood cells in the blood, which in turn leads to various health problems, such as anaemia, infections or increased risk of bleeding.
The impaired maturation of the blood stem cells also results in a continuous increase of immature blood cells (so-called blasts) in the bone marrow. Because of their immaturity, they are unable to fulfill their assigned functions. In some MDS patients, the amount of blasts in the bone marrow increases to a degree that does not allow to differentiate MDS from a leukaemia any more. Hence, MDS was formerly also known as “pre-leukaemia”.
Incidence
While myelodysplastic syndromes (MDS) are the most frequent malignant diseases of the bone marrow in older people, it is very rare in childhood and adolescence. MDS accounts for about 8 % of all blood cancer diseases in children and adolescents under 18 years of age and for about 2.5 % of all cancers in this age group.
According to the German Childhood Cancer Registry, about 54 children and adolescents aged between 0 and 17 years are newly diagnosed with MDS in Germany every year. The young patients’ average age at diagnosis is approximately nine years. Boys are slightly more affected than girls (sex ratio: 1,2 : 1).
Types of myelodysplastic syndrome (MDS)
Depending on the way a myelodysplastic syndrome (MDS) has developed and based on microscopical features of blood and bone marrow, there are different types of MDS, which will be introduced in the following paragraphs.
Primary and secondary MDS
Myelodysplastic syndrome (MDS) often develops without an obvious reason. It’s known as “primary” MDS. About 75 % of children with MDS are in this group. t is assumed, however, that „primary“ childhood MDS may also involve genetic alterations that have not been identified yet (see chapter „Causes“).
In some patients, development of MDS may be associated with certain triggers; those cases are called “secondary” MDS. For example, some patients develop MDS after a preceding radiation therapy or chemotherapy done for treatment of another, often malignant disease. Others have been suffering of an underlying congenital disease before the diagnosis of MDS. These include, for example, Fanconi anaemia, dyskeratosis congenita, Shwachman-Diamond syndrome, Diamond-Blackfan anaemia (DBA) or a severe congenital neutropenia, all of which involve a dysfunction of the bone marrow. Also, severe aplastic anaemia (SAA) may precede the diagnosis of MDS.
Subtypes of primary childhood MDS as per WHO classification
According to the classification of the World Health Organization (see WHO classification), primary childhood MDS is subdivided into two subtypes. This differentiation is mainly based on the blast counts in blood and bone marrow.
Refractory cytopenia of childhood (RCC)
The blast count in patients with „refractory cytopenia of childhood“ (RCC) is usually below 2 % in the peripheral blood and below 5 % in the bone marrow and thereby not much higher than in healthy children. However, the bone marrow in children with RCC is frequently very low on cells, which means that the numbers of all the cells usually found in the bone marrow (red and white blood cells, platelets) are reduced. Therefore, another congenital bone marrow disease (bone marrow failure) or aplastic anaemia always need to be considered as a differential diagnosis, because these conditions typically present with low bone marrow cell counts as well.
Myelodysplastic syndrome with excess blasts (MDS-EB)
In MDS patients, blast counts [see blasts] may markedly exceed 2 % or 5 %, respectively. This type is known as MDS-EB, meaning MDS with blast excess. However, bone marrow blast counts should not exceed 29 %, because then it would be called a leukaemia. Since other factors are also being considered when differentiating between MDS and leukaemia, such as the rate of blast increase, distinction (differential diagnosis) between MDS and leukaemia can sometimes be challenging. In borderline scenarios, such as dealing with blast counts between 20 and 30 %, it is recommended to repeat diagnostics by bone marrow puncture after a couple of weeks to confirm diagnosis.
|
MDS Subtype |
Blood |
Bone marrow |
|---|---|---|
|
Refractory cytopenia of childhood (RCC) |
less than 2 % blasts |
less than 5 % blasts |
|
Myelodysplastic syndrome with blast excess (MDS-EB) |
2 – 29 % blasts |
5 – 29 % blasts |
Causes
The exact causes for myelodysplastic syndrome (MDS) mostly remain unclear; however, like other types of cancer, the disease is neither contagious nor can it be transferred to other humans. In most cases, MDS develops in previously healthy children without an apparent reason (so-called primary MDS). It is assumed, however, that the affected have a certain predisposition for development of an MDS or a leukaemia, respectively.
Cause of the predisposition are altered genes (mutations) that are also present in sperm or egg cells (hence in the germline) and can therefore be inherited. In this case, the mutation is present in all body cells of the patient, too. The affected children have inherited this germline mutation from either one or both parents, or the gene alteration has developed de novo (sponateously) in the fertilized egg cell.
It has been known for many years that gene alterations that are capable of causing impaired haematopoiesis in the bone marrow (such as Fanconi anaemia, severe congenital neutropenia, dyskeratosis congenita or Diamond-Blackfan anaemia) are also associated with a predisposition for MDS. Over the past years, additional inheritable diseases that play a role have been identified. These include the so-called GATA2 deficiency or the SAMD9/SAMD9L syndrome. Since these congenital diseases are usually associated with an increased cancer risk, they are also known as cancer predisposition syndromes.
MDS develops differently in older persons. Most adults do not present with congenital impairments, but with gene alterations acquired over decades, which can transform a cell slowly into an MDS or leukaemia cell. Some patients have received chemotherapy or radiation therapy (radiotherapy) for another malignancy prior to the diagnosis of MDS. In those cases. MDS thus represents the secondary disease (secondary MDS), which has at least partially been induced by the treatment of the primary malignancy.
Symptoms
Symptoms in a patient with myelodysplatic syndrome (MDS) are mainly determined by the severity of the lack of functioning blood cells, meaning the degree of cytopenia. Depending on which blood cells are affected, the following types of cytopenia and associated symptoms can occur:
Anaemia: lack of red blood cells
The job of the red blood cells (erythrocytes) is to transport the oxygen, which is inhaled by the lungs, to the different organs and tissues of the body. Lack of red blood cells (anaemia) leads to pallor, fatigue, weakness and headache.
Immune deficiency: lack of white blood cells (leukopenia, neutropenia)
White blood cells (leukocytes) are responsible for the defense of pathogens and hence for the prevention of infections. There are different types of leucocytes, for example lymphocytes and granulocytes, which have different functions in immune defence. In case of lack of functioning white blood cells, so-called leukopenia, the body is at risk of infection. Frequently, patients with MDS in particular present with reduced granulocyte count (so-called granulocytopenia or neutropenia). Since these are responsible for fighting bacteria and fungi, MDS patients often have bacterial or fungal infections associated with fever.
Bleeding tendency: lack of platelets (thrombocytopenia)
Platelets (thrombocytes) play an important role in clotting. If their count is reduced (so-called thrombocytopenia), the risk of both spontaneous and injury-induced bleeds increases. These may, for example, present as small red spots on the skin or mucous membranes (petechiae), bruises (haematoma) and/or nose-/gum bleeds, but sometimes also as severe bleedings in inner organs or the brain. The risk of severe bleeds increases with the degree of thrombocytopenia.
MDS patients with blast excess (MDS-EB, see chapter „Types of MDS“) sometimes present with side effects that are difficult to treat, such as inflammation of small blood vessels or the so-called Sweet syndrome, which is characterized by fever and red nodules on the skin.
Diagnosis
If the doctor, based on the young patient’s history (anamnesis) and physical examination, suspects a blood disease, he or she will first initiate a comprehensive blood test. If the results, due to certain blood count changes, promote the diagnosis of a blood disease such as myelodysblastic syndrome (MDS), a sample of the bone marrow (bone marrow biopsy) is required for confirmation. For bone marrow tests and other diagnostic procedures, the doctor will refer the patient to a children's hospital with a paediatric oncology program (paediatric oncology unit).
Analysis of blood and bone marrow
Since the symptoms in MDS patients are not specific for MDS and can also present in other blood diseases such as leukaemias, diagnosis can only be confirmed by comprehensive analysis of the blood and the bone marrow.
Bone marrow obtained from the pelvic bone is required for establishing the final diagnosis of MDS. For this, bone marrow puncture and bone marrow punch biopsy are necessary. Both methods add to one another and are done during the same procedure. For bone marrow aspiration, a special syringe is used to aspirate a small amount of bone marrow. A bone marrow biopsy is performed by using a larger syringe to take larger samples of marrow (about 1 mm in diameter). Further information on bone marrow puncture and punch biopsy can be found here.
After obtaining the sample, the blood forming bone marrow cells are examined under the microscope by a specialist for blood and malignant diseases. In MDS, cells may look different in various ways (dysplasia), which are crucial for diagnosis. In MDS with blast excess (MDS-EB), bone marrow blast counts are increased (see chapter “Types of MDS”). The sample obtained by bone marrow aspiration is also used for chromosomal analysis (cytogenetics, see below). If MDS is the suspected diagnosis, bone marrow puncture and punch biopsy should be repeated after about 14 days in order to securely confirm the diagnosis.
Examination of the chromosomes (cytogenetics)
Chromosomal changes in the bone marrow and blood cells are found in more than half of the children with MDS with excess blasts (MDS-EB) and in about 30 % of patients with refractory cytopenia (RCC). Identification of these changes helps with establishing the diagnosis of MDS. The most common and typical chromosomal aberration in children with MDS is the loss of chromosome 7 (so-called monosomy 7). In these cases, cells only have one chromosome 7 instead of two.
Molecular genetic testing
Some children and adolescents with MDS present with a genetic predisposition for the disease, meaning that they have a germline mutation (see chapter “Causes”). For the analysis of these genetic alterations, cells other than blood and/or bone marrow, such as hair follicle cells (germline material), require molecular genetic analysis as well. In case of identification of a germline mutation in a patient, a potentially matching family donor for stem cell transplantation (SCT) also needs to be tested for this mutation. Only a family member without this mutation should consider to serve as a donor.
Treatment
Treatment of patients with myelodysplastic syndrome (MDS) depends on the form of the disease, meaning, on whether it is a primary or secondary MDS or, in the first case, whether it is refractory cytopenia of childhood (RCC) or an MDS with excess blasts (MDS-EB), respectively (see chapter „Types of MDS“). Treatment basically consists of the following options:
- Transplantation of blood-forming stem cells from a donor (allogeneic stem cell transplantation, SCT) following a conditioning therapy (high-dose chemotherapy)
- immunosuppressive therapy (IST)
- other kinds of drug-based therapies: chemotherapy, agent azacitidin
- supportive treatments: blood transfusions, antibiotic therapy [see antibiotics]
The following paragraphs will introduce treatment of patients with different types of MDS.
Refractory childhood cytopenia (RCC)
Patients with refractory childhood cytopenia (RCC) receive – depending on presence or absence of chromosomal aberrations, extent of lack of blood cells (cytopenia) and on cell content (cellularity) in the bone marrow – different therapies.
RCC patients presenting with only one chromosome 7 (monosomy 7), as diagnosed by chromosomal analysis, usually experience a less favourable course of the disease with a risk of blast increase within one to two years. Hence, these patients should receive allogeneic stem cell transplantation (SCT) as soon as possible. For this form of treatment, patients first receive high doses of cytostatics (high-dose chemotherapy) as the so-called conditioning regimen, in order to eliminate all (healthy and sick) bone marrow cells. After that, the recipient receives healthy blood stem cells obtained from the bone marrow or peripheral blood of a donor.
Patients with RCC without chromosomal aberrations may experience stable disease for years. If they don’t require any blood transfusions and present with sufficient granulocytes (white blood cells that defend bacteria and fungi), they won’t be treated at first and just observed, with the course of the disease being monitored via regular (blood) tests (so-called watch-and-wait strategy). If the results of the blood tests do deteriorate over time, treatment, usually stem cell transplantation, may be indicated after all.
Patients with normal chromosomal findings who require blood transfusions shortly after diagnosis or who present with very low granulocyte counts will receive stem cell transplantation as soon as possible. Healthy siblings or a matching unrelated donor are chosen for stem cell donation. In case of no matching sibling, immunosuppressive therapy (IST) is the gold standard for patients with hypocellular bone marrow. This immunosuppressive therapy is similar to treatment of aplastic anaemia (severe aplastic anaemia, SAA) and can take many months or years.
With immunosuppressive therapy (IST), sufficient increase of blood cell counts can be achieved in about half of the patients, so that blood transfusions are not required any longer. It is also possible that cytopenia recurs after an initially successful IST (so-called recurrence). In case of recurrent disease or non-responding to IST, stem cell transplantation from an appropriately matching non-related donor is recommended.
MDS with excess of blasts (MDS-EB)
MDS patients with excess of blasts (MDS-EB), considered as an advanced stage of the disease, have a high risk of developing a leukaemia later. Hence, early stem cell transplantation is the therapy of choice for MDS-EB. Healthy siblings or a matching unrelated donor are options for stem cell donation. In case of very high blast counts [see blasts], pretreatment with chemotherapy (azacitidin treatment) for blast reduction may be indicated prior to conditioning (high-dose therapy) and stem cell transplantation.
Secondary MDS
Patients with a secondary myelodysplastic syndrome (MDS) are usually treated like patients with primary MDS with excess blasts (MDS-EB) (see previous chapter). Hence, these patients should receive stem cells from a matching donor shortly after diagnosis.
Supportive therapy during treatment
In all patients with myelodysplastic syndrome (MDS), supportive treatment measures (supportive therapy) are appropriate and required. These supportive measures help to reduce symptoms caused by the disease and to manage or prevent treatment-induced side-effects.
Most patients present with anaemia and/or lack of platelets (thrombocytopenia) at diagnosis, which are associated with health problems (see chapter „Symptoms“). These problems can be treated by blood transfusions (transfusion of red blood cells or platelets, respectively). However, repetitive transfusions of red blood cells are associated with high loads of iron, which, over time, can deposit in organs (in particular liver and heart) and harm them (so-called iron overload, haemochromatosis). Therefore, patients with iron overload require iron deprivation treatment.
Patients‘ immune defence mechanisms are weakened by the MDS itself and also by its therapy, such as stem cell transplantation or immunosuppressive therapy. Hence, they need to be protected from infections as best as possible and, in case of infection, treated as soon as possible. Infections in immunosuppressed children should always be considered as life-threatening.
Fever is usually the first symptom of an infection. Parents or relatives with a fever should, therefore, immediately contact the caregiver team (even at night), so that the children can receive broad spectrum antibiotics as soon as possible. In patients with neutropenia (lack of granulocytes), preventive treatment with antibiotics and antimycotics (agents against fungi) is indicated.
For further details on this topic, please see our information on "supportive care".
Prognosis
Myelodysplastic syndrome (MDS) can have different courses. Some diseases remain stable over a long period of time, while others progress rapidly. A patient’s prognosis is dependent on the subtype of the disease (see chapter „Types of MDS“) and on the presence of genetic alterations in the bone marrow cells.
Prognosis of patients with refractory childhood cytopenia (RCC)
Patients with refractory childhood cytopenia (RCC) are – based on the presence or absence of certain risk factors – assigned to different treatment groups, thereby receiving different therapies (observation, stem cell transplantation or immunosuppressive therapy, see chapter “Treatment”). Overall, these patients have a favourable prognosis with a probability of survival of 80 – 90 %, regardless of the treatment group. This also applies to RCC patients with monosomy 7, who have received allogeneic stem cell transplantation early due to the high risk of advanced MDS or leukaemia (progressive disease). The prognosis with such a treatment is as favourable as that of other RCC patients.
In RCC, the risk of relapse (recurrence) after stem cell transplantation is very low. However, every stem cell transplantation is an intensive therapy, involving substitution of the ill bone marrow by a healthy bone marrow of a donor following high-dose chemotherapy. Hence, these patients should make at least yearly follow-up appointments in order to have long-term sequelae diagnosed and treated in a timely manner.
Patients who have been successfully treated with immunosuppressive therapy (IST) or who are being observed without treatment, also require regular bloodwork as well as a bone marrow punch biopsy and cytogenetic testing once a year in order to diagnose recurrent or progressive disease as early as possible.
Prognosis of patients with advanced MDS (MDS-EB) or secondary MDS
Allogeneic stem cell transplant has proven to be a curative treatment option for children and adolescents with MDS with excess blasts (MDS-EB) or with secondary MDS. About 50 – 60 % of patients can be cured by this therapy. Patients with severe chromosomal aberrations, such as three or more chromosomal changes (so-called complex karyotype), have a rather unfavourable prognosis.
 PDF - Brief information on Myelodysplastic Sydrome (MDS) (303KB)
PDF - Brief information on Myelodysplastic Sydrome (MDS) (303KB)
05/09/2023, glossary added: 10/03/2024
References 
- Rudelius M, Weinberg OK, Niemeyer CM, Shimamura A, Calvo KR: The International Consensus Classification (ICC) of hematologic neoplasms with germline predisposition, pediatric myelodysplastic syndrome, and juvenile myelomonocytic leukemia. Virchows Archiv : an international journal of pathology 2023, 482: 113 [PMID: 36445482]
- Bortnick R, Wlodarski M, de Haas V, De Moerloose B, Dworzak M, Hasle H, Masetti R, Starý J, Turkiewicz D, Ussowicz M, Kozyra E, Albert M, Bader P, Bordon V, Cario G, Beier R, Schulte J, Bresters D, Müller I, Pichler H, Sedlacek P, Sauer MG, Zecca M, Göhring G, Yoshimi A, Noellke P, Erlacher M, Locatelli F, Niemeyer CM, Strahm B, for EWOG-MDS: Hematopoietic stem cell transplantation in children and adolescents with GATA2-related myelodysplastic syndrome. Bone marrow transplantation 2021, E-pub ahead of printg [PMID: 34244664]
- Erdmann F, Kaatsch P, Grabow D, Spix C: German Childhood Cancer Registry - Annual Report 2019 (1980-2018). Institute of Medical Biostatistics, Epidemiology and Informatics (IMBEI) at the University Medical Center of the Johannes Gutenberg University Mainz 2020 [URI: https://www.kinderkrebsregister.de/ typo3temp/ secure_downloads/ 42507/ 0/ 1c5976c2ab8af5b6b388149df7182582a4cd6a39/ Buch_DKKR_Jahresbericht_2019_komplett.pdf]
- Niemeyer C, Kratz C: Myelodysplastische Syndrome. in Niemeyer C, Eggert A (Hrsg): Pädiatrische Onkologie und Hämatologie 2018, Springer Verlag; 715 [ISBN: 978-3-662-43685-1]
- Niemeyer C, Eggert A (Hrsg): Pädiatrische Hämatologie und Onkologie. Springer-Verlag GmbH Deutschland 2. vollständig überarbeitete Auflage 2018 [ISBN: 978-3-662-43685-1]
- Ripperger T, Bielack SS, Borkhardt A, Brecht IB, Burkhardt B, Calaminus G, Debatin KM, Deubzer H, Dirksen U, Eckert C, Eggert A, Erlacher M, Fleischhack G, Frühwald MC, Gnekow A, Goehring G, Graf N, Hanenberg H, Hauer J, Hero B, Hettmer S, von Hoff K, Horstmann M, Hoyer J, Illig T, Kaatsch P, Kappler R, Kerl K, Klingebiel T, Kontny U, Kordes U, Körholz D, Koscielniak E, Kramm CM, Kuhlen M, Kulozik AE, Lamottke B, Leuschner I, Lohmann DR, Meinhardt A, Metzler M, Meyer LH, Moser O, Nathrath M, Niemeyer CM, Nustede R, Pajtler KW, Paret C, Rasche M, Reinhardt D, Rieß O, Russo A, Rutkowski S, Schlegelberger B, Schneider D, Schneppenheim R, Schrappe M, Schroeder C, von Schweinitz D, Simon T, Sparber-Sauer M, Spix C, Stanulla M, Steinemann D, Strahm B, Temming P, Thomay K, von Bueren AO, Vorwerk P, Witt O, Wlodarski M, Wössmann W, Zenker M, Zimmermann S, Pfister SM, Kratz CP: Childhood cancer predisposition syndromes-A concise review and recommendations by the Cancer Predisposition Working Group of the Society for Pediatric Oncology and Hematology. American journal of medical genetics. Part A 2017, 173: 1017 [PMID: 28168833]
- Yoshimi A, Strahm B, Baumann I, Furlan I, Schwarz S, Teigler-Schlegel A, Walther JU, Schlegelberger B, Göhring G, Nöllke P, Führer M, Niemeyer CM: Hematopoietic stem cell transplantation in children and young adults with secondary myelodysplastic syndrome and acute myelogenous leukemia after aplastic anemia. Biology of blood and marrow transplantation 2014, 20: 425 [PMID: 24316460]
- Niemeyer C: Myelodysplastic syndrome and aplastic anemia in children. Europ School of Oncology 2000, 31