Ewing sarcoma (brief information)
A Ewing sarcoma is a rare malignant tumour in childhood and adolescence that originates mostly in the bones. This text provides information about the characteristics of this disease, its frequencies, causes, symptoms, diagnosis, treatment, and prognosis.
Author: Maria Yiallouros, Editor: Maria Yiallouros, Reviewer: Prof. Dr. med. Uta Dirksen, , Virginia Santarini, Dr. med. Naghmeh Niktoreh, English Translation: Dr. med. Gesche Riabowol (geb. Tallen), Last modification: 2026/06/26 https://dx.doi.org/10.1591/poh.patinfo.ewing.kurz.20101215
Table of contents
General information on the disease
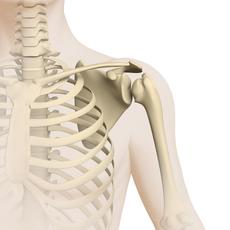
Ewing sarcomas are malignant, solid tumours that mostly arise within the bone tissue. The disease is named after the American cancer researcher Dr. James Ewing (1866-1943), who first described this tumour in 1921. Most Ewing’s sarcomas grow and spread very quickly. Without an appropriate treatment, the disease is always fatal.
In general, Ewing sarcoma can develop in any bone. However, pelvic bones, followed by the long bones of the upper or lower leg, the thoracic skeleton, the plate bones, and the spine are those most frequently affected.
The tumour may spread (metastasize) both within the bone and into the adjacent soft tissue. Rarely (about 15–20 %), Ewing sarcoma arises directly within soft tissue, that is to say, outside the bone and without the bone being involved. Such tumours are called extrasceletal or extraosseal Ewing sarcomas. Ewing sarcomas of pure soft tissue origin may be located in the kidneys, adrenal glands, lungs, or in the gastrointestinal tract.
Due to the rapid growth and spread of Ewing sarcomas, about 25 % of the children and adolescents with this disease already present with detectable daughter tumours (metastases) at the time of diagnosis, primarily in the lungs, but also in the bones and, less often, in the bone marrow. It has to be assumed for all other patients, too, that tumour cells have already spread elsewhere via the blood or lymphatic stream and, thus, have formed tiny metastases in other organs. These so-called micrometastases may not be detected at diagnosis due to their small size. Since Ewing sarcoma is thus considered a disease that may affect the whole body, it is also called a systemic disease.
Incidence
Following osteosarcomas, Ewing sarcomas are the second most common primary bone tumours in childhood and adolescence; they account for about 2 % of all pediatric malignancies. According to the German Childhood Cancer Registry (Mainz, Germany), approximately 50 children and adolescents under the age of 18 (about 3 of 1,000,000) are diagnosed with Ewing sarcoma in Germany per year.
Ewing sarcoma may develop at any age; however, the frequency of this type of cancer peaks in the second decade of life. In childhood and adolescence (age group 0–17 years), the median age at diagnosis is 13 years, with adolescents between 12 and 17 years of age being the most frequently affected. Nevertheless, Ewing sarcomas are also observed in babies, infants, and school children. Overall, boys and male adolescents are more frequently affected by the disease than girls (gender ratio: 1.5:1).
Histological characteristics and tumour types
Ewing sarcomas are primitive malignant tumours. It is still unknown as of today, from which precursor cell they arise. According to the most recent research, they develop from immature (undifferentiated) tissue cells, so-called mesenchymal stem cells, or from primitive neuroectodermal stem cells.
Ewing sarcoma cells are histologically assigned to the mesenchymal small-, blue- and round cell tumour types; they can only be differentiated from similar looking, undifferentiated tumour cells of other malignancies (such as neuroblastoma, medulloblastoma, Non-Hodgkin lymphoma, soft tissue sarcoma and small-cell osteosarcoma) by immunohistochemical and molecular genetic analyses. Ewing sarcomas are characterised by specific gene fusions, most commonly a fusion of the EWSR1 and FLI1 genes. Detecting this genetic alteration is now an important part of the diagnostic process. Since these tumours are rare, the associated analyses are carried out in specialised laboratories (see chapter “Diagnosis”).
Until recently, different tumour types were distinguished within the group of Ewing sarcomas on the basis of their histological features and the site of origin of the tumour. These included the classic Ewing sarcoma (EWS), the peripheral malignant primitive neuroectodermal tumour (PPNET or pPNET), the Askin tumour of the chest wall and Ewing tumours of soft tissues. According to the World Health Organization's current classification (WHO classification), all these tumours are summarised as Ewing sarcomas on the basis of their shared characteristic genetic alterations. All Ewing sarcomas are highly malignant tumours.
Causes
The underlying causes for the development of a Ewing sarcoma are still unknown. Neither external factors, such as a previous radiation therapy, nor inherited genetic factors (genetic predisposition) seem to play a relevant role. However, the disease shows ethnical preference by developing more frequently in white-skinned people (Caucasians) than in people of colour (Asians or Africans).
It is also known that tumour cells of Ewing sarcomas present with certain chromosomal aberrations, which all include a certain gene on chromosome 22 – the so-called Ewing sarcoma gene (EWS gene). These aberrations develop due to an exchange of chromosomal parts (translocation), mostly between the EWS gene on chromosome 22 and a gene on chromosome 11 (particularly FLI1). Accounting for 85 %, the most frequent translocation (so-called [t(11;22) (q24;q12) translocation]) results in the EWSR1 and FLI1 genes fusing together. This gene fusion is so specific for Ewing sarcoma that it allows confirmation of the diagnosis.
For many Ewing sarcomas, additional genetic alterations have been identified (for example increased numbers of chromosomes or loss or gain of genetic material, respectively). The genetic defects resulting from those aberrations promote that a healthy cell transforms into a tumour cell. In general, those genetic defects, which are found in tumour tissue only, are not inherited.
Ewing sarcoma very rarely occurs in association with a cancer predisposition syndrome – a hereditary predisposition to developing tumours – or as a secondary cancer (secondary malignancy). The latter may occur even many years after treatment for an initial (primary) cancer has been completed.
Symptoms
The most frequent symptoms of Ewing sarcoma are pain and a swelling in the tumour region.
Pain may be intermittant and is usually activity-dependent, but frequently also occurs at rest, for example during night time. With increasing tumour growth, the pain may be accompanied by a visible and/or palpable lump at the tumour site. The swelling may be reddened and may sometimes lead to an impaired mobility, which initially may be mistaken for growing pains or a sports injury or some bone inflammation.
Since Ewing sarcomas may develop in any bone as well as in soft tissue, further symptoms are quite diverse, depending on the site and extent of the tumour. In case the spinal column and/or peripheral nerves (peripheral nervous system) are affected, deficits such as paralysis may be the major symptom. Tumours in the pelvic or chest region or in the thigh may remain unidentified for quite a while. About one third of patients complain of general symptoms such as fever, feeling of illness (malaise), weight loss and/or fatigue, which may be indicative of an advanced disease. For some patients, only a few weeks, for others up to several months may pass between first symptoms and the diagnosis of osteosarcoma.
Good to know: Not all children and adolescents presenting with the complaints described above suffer from Ewing sarcoma or any other malignant bone tumour. However, every type of musculoskeletal pain in a child or a teenager should be taken seriously and be dealt with by an experienced paediatrician in order to appropriately rule out an underlying cancer.
Diagnosis
If the paediatrician thinks that the young patient’s history (anamnesis) and physical examination are suspicious of a malignant bone tumour, the child should immediately be referred to a hospital with a childhood cancer program (paediatric oncology unit), where further diagnostics can be initiated and performed by childhood cancer professionals. Ideally, the hospital is part of a specialist sarcoma centre or collaborates with one.
Very close collaboration between various specialists (such as paediatric oncologists, paediatric surgeons, paediatric radiologists, to name a few) is required both to find out whether the patient really suffers from a malignant bone tumour and, if so, to determine the tumour type and the extension of the disease. Knowing these details is crucial for optimal treatment planning and prognosis.
Laboratory tests
The diagnosis of Ewing sarcoma requires, aside from a comprehensively taken history and physical exam, blood and urine tests. Although tumour markers that are specific for Ewing sarcoma have not been identified yet, certain peculiarities indicative of the type of the disease and/or differential diagnoses do exist, and can be discovered via laboratory testing.
Imaging tests to confirm tumour existence
Based on their typical radiological features, most malignant bone tumours can already be diagnosed by X-ray examination. Additional imaging procedures such as magnetic resonance imaging (MRI) and/or computed tomography (CT) with a contrast agent subsequently help to define the exact tumour site and size as well as its demarcation with regard to adjacent tissue (such as blood vessels, muscles, tendons or joints). Nearby metastases (called skip metastases) are easily detectable by these methods as well.
For imaging of affected soft tissue and bone marrow, MRI is superior to CT. Apart from plain x-rays, it is, therefore, considered as the gold standard in radiological diagnosis of a Ewing sarcoma. It also serves as a basis for planning subsequent surgery and for monitoring the course of the disease during chemotherapy. However, a CT may be additionally necessary in order to examine bone changes more closely.
Obtaining a tumour sample (biopsy)
For final confirmation of the diagnosis, tumour tissue is required to be removed (biopsy). The biopsy should only be performed by doctors who are specialized in surgery of sarcomas. This ensures that the access chosen for biopsy does not cause problems for subsequent treatment. An unfavourably planned biopsy may result in the requirement of a far more extensive subsequent surgery than initially necessary or, worst case scenario, in the inoperability of a tumour that would have been initially removable.
The required tissue samples are either gained during the surgical procedure for tumour resection (open surgery) or by needle biopsy. The latter uses special needles to punch multiple samples of tumour tissue. The obtained samples are subsequently analysed histologically, immunohistochemically and molecular genetically by specialists. Molecular genetic analysis has special relevance, since identification of genetic alterations that are typical for Ewing sarcomas confirm the diagnosis and allow ruling out other, similar tumour types (see chapter „Causes“).
Tests to assess tumour spread (staging)
Once the diagnosis of a Ewing sarcoma has been confirmed, further tests are performed to assess potential tumour spread into other organs (metastasis). In this context, diagnostic imaging plays a major role. At first, accurate measuring of the primary tumour is required (so-called volumetry), since its volume (and shrinking during treatment) has a major impact on the patient’s probability of survival.
In order to assess or, respectively, rule out lung metastases, a computed tomography of the chest (chest-CT) is performed. A skeletal scintigraphy (bone scan) using radioactively labelled technetium (99mTc) previously served to detect or rule out bone metastases. Today, positron emission tomography (PET) with radioactively labelled glucose (18F-fluoride-deoxyglucose, short FDG) is performed instead of a bone scan. This very sensitive nuclear medical imaging procedure is either combined with magnetic resonance imaging or computed tomography. Regardless of whether bone scan or PET are chosen, an MRI is also done for all clinically and radiologically suspicious regions. Some patients benefit from a whole-body MRI.
To find out whether the bone marrow is affected, tissue samples are obtained by bone marrow puncture and bone marrow punch biopsy and, subsequently, analysed histologically and molecular genetically. If the central nervous system (brain and spinal cord) is suspected to be affected as well, it may even be necessary to take a sample of cerebrospinal fluid (lumbar puncture).
Tests before treatment begins
Before treatment begins, further tests are needed in order to assess the condition of different organs. Therefore, the doctors will recommend an electrocardiography (ECG) as well as an ultrasound of the heart (echocardiography), a hearing test (by means of audiometry, BERA hearing test and otoacoustic emissions), special diagnostics for determining kidney and lung functions as well as various blood tests. Any changes occurring during the course of treatment can be assessed and managed better based on the results of those initial tests, which thus help to keep the risk of certain treatment-related side effects as low as possible.
In addition, patients should be advised on fertility and fertility preservation, and, where possible, measures to preserve fertility should be taken: For male and fema-le patients of reproductive age, retrieval and freezing (cryopreservation) of sperm or one (or part of) an ovary, respectively, should be considered.
Good to know: Not all of the above-mentioned tests will be done for every single patient. On the other hand, additional tests not mentioned here may be required individually. Ask the doctor which diagnostics are necessary and why.
Treatment planning
After the diagnosis has been confirmed, therapy is planned. In order to design a highly individual, risk-adapted treatment regimen for the patient, certain individual factors influencing the patient’s prognosis (called risk factors or prognostic factors) are being considered before and during treatment (risk-adapted treatment strategy).
Important prognostic factors are the type, localisation, size and spread of the tumour (local or metastasised), which are assessed before treatment begins. In addition, the extent of surgical tumour/metastasis removal (imcomplete versus complete) as well as the response of the disease to chemotherapy have major impact on treatment planning. All these factors are included in treatment planning in order to achieve the best outcome possible for each patient.
Treatment
Treatment of children and adolescents with Ewing sarcoma should take place in a children's hospital with a paediatric oncology program. Only in such a childhood cancer centre, highly experienced and qualified staff (doctors, nurses and many more) is guaranteed, since they are specialized and focus on the diagnostics and treatment of children and teenagers with cancer according to the most advanced treatment concepts. The doctors in these centres collaborate closely with each other. Together, they treat their patients according to treatment plans (protocols) that are continuously optimised. Treatment in a centre with assigned sarcoma focus is considered optimal. Alternatively, the paediatric oncology program should collaborate with such a centre, in particular regarding surgery and radiotherapy.
The goal of the treatment is to achieve higher cure rates while avoiding side effects as much as possible.
Treatment methods
Treatment of children and adolescents with Ewing sarcoma consists of surgery and/or radiotherapy (local therapy) as well as chemotherapy. For some patients, a high-dose chemotherapy followed by autologous stem cell transplantation may be an option, too.
Radiation therapy (radiotherapy) is done using energy-rich, electromagnetic radiation (photons oder protons), given through the skin to the tumour region. Radiation causes DNA damage in tumour cells, thereby leading to cell death. Chemotherapy uses drugs (so-called cytostatic agents or cytostatics) that can kill fast-dividing cells, such as cancer cells, or inhibit their growth, respectively.
Surgery and radiotherapy aim at maximally possible local disease control. Additional chemotherapy is necessary, because it appeared that though surgery and radiotherapy alone are capable of eliminating the tumour, metastases will almost always develop later on. Therefore a treatment is required – such as chemotherapy – that affects the whole body (so-called systemic treatment). However, chemotherapy alone cannot replace local therapy.
In certain patients with locally confined (localised) disease and an unfavourable response to induction chemotherapy, high-dose chemotherapy followed by autologous stem cell transplantation may be considered. Otherwise, no benefit of this form of treatment over standard therapy has been demonstrated in patients receiving first-line treatment.
Course of treatment
Usually, local therapy is provided between two cycles of chemotherapy. Overall, treatment takes about ten months, but – depending on many different factors – may also require more time. The different treatment phases will be outlined in the next paragraphs.
Chemotherapy prior to local therapy
Therapy is generally initiated by an intensive chemotherapy of several weeks (called induction chemotherapy or induction therapy). The goal of this chemotherapy is to shrink the tumour and potential spread (metastases) in order to optimise the conditions for subsequent surgical tumour removal, thereby contributing to safety and efficacy of surgery. Also, chemotherapy helps eliminate micrometastases (the tiny metastases that cannot be detected by imaging diagnostics), thus preventing further tumour spread.
In order to eliminate as many tumour cells as possible, patients receive a combination of different chemotherapeutic agents (cytostatics) that have proven to be effective in the treatment of Ewing sarcoma.
The standard combinations of cytostatic drugs currently in use include vincristine, doxorubicin and cyclophosphamide (VDC), as well as ifosfamide and etoposide (IE), which are given in alternation. The VDC/IE combinations are administered by infusion over a total of nine chemotherapy cycles, each lasting several days, during which the patient needs to be inpatient. Between courses, patients can go home as outpatients. Readmission is only required in case of severe side-effects.
Local therapy – surgery and radiotherapy
Still during or latest after cessation of chemotherapy, local therapy is scheduled. It is an essential part of the treatment for Ewing sarcoma and includes surgical and radiotherapy options. Where possible, the aim is to remove the tumour completely by surgery. Depending on the tumour’s location, the extent of the disease and the response to chemotherapy, radiotherapy may also be required. In some cases, local treatment consists solely of surgery or solely of radiotherapy.
Which one of the two treatment methods is indicated or whether they will be combined depends on the patient and his / her disease condition and, thus, needs to be discussed on a case-by-case basis within an interdisciplinary team. Your caregiver team will inform you more in detail on the type and course of surgery or radiotherapy, respectively.
Surgery
If surgery is carried out, the aim is to remove the tumour as completely as possible; therefore, the surgical procedure must be performed at a specialist sarcoma centre.
Thanks to major advances in limb-preserving surgical techniques and the use of chemotherapy and/or radiotherapy, it is frequently possible as of today to waive amputation for patients with large tumours of the arms or legs. Different techniques are available for limb reconstruction based on the location and the size of the tumour. For example, metal joint implants are frequently used; for children, also growing endoprotheses are available.
Following surgery, a pathologist examines the tumour tissue under the microscope to find out how the disease has responded to the preceding chemotherapy. This so-called histological response is assessed by measuring the amount of the tumour cells that are still alive. A good response to treatment is generally associated with a more favourable prognosis and is taken into account in the planning of further treatment. In addition to tumour response, however, treatment planning considers other factors as well, such as the extent of the disease, the presence of metastases and the outcome of the operation.
Good to know: As far as possible, metastases identified at the time of diagnosis are treated locally like the primary tumour, thus being surgically removed and/or irradiated.
Radiotherapy
Ewing sarcoma is sensitive to radiotherapy. Radiation can generally be administered before or after surgery, or as a stand-alone treatment. The latter is usually only an option if complete tumour removal is not possible or would involve significant risks or functional impairment.
The tumour region, plus a safety margin, is irradiated. The total radiation dose depends, amongst other things, on the location of the tumour and on whether radiotherapy is used as part of a combination treatment or as a stand-alone local therapy. The total dose is usually between 45 and 68.4 gray (Gy) and is determined on an individual basis. The optimal radiation dose is currently being investigated as part of the ongoing treatment optimising trial (see chapter "Therapy optimising trials and registries“).
Chemotherapy after local therapy
After local therapy,chemotherapy is continued. This phase of treatment is known as consolidation chemotherapy or consolidation therapy and is designed to eliminate any remaining tumour cells in the body and further reduce the risk of relapse. The standard consolidation therapy for all patients consists of the two combination chemotherapy regimens ifosfamide/etoposide (IE) and vincristine/cyclophosphamide (VC), which are administered alternately. A total of five cycles of chemotherapy are given.
In patients with lung metastases, radiotherapy of the entire lung may also be considered following significant tumour regression. For patients with metastases outside the lung, treatment is tailored to the individual’s clinical situation and is determined by the multidisciplinary treatment team.
Good to know: in some cases, follow-up radiotherapy may be carried out after chemotherapy. In order to prevent or adequately manage the side effects of the intensive therapy, specific supportive care (supportive therapy) regimens have been established. Here you will find information on supportive care as well as recommendations for home, the latter of which may be helpful during or after chemo- and radiotherapy.
Treatment in case of progressive or recurrent disease
Despite optimised treatment methods, some patients with Ewing sarcoma will experience relapse of the disease (recurrence). There is no generally applicable standard treatment recommendation for these patients. Treatment is tailored to the individual’s clinical situation and may include multi-agent chemotherapy (for example with topoisomerase inhibitors such as etoposide, irinotecan or topotecan, or alkylating agents (alkylants) like ifosfamide, cyclophosphamide and temozolomide), radiotherapy, surgery or a combination of these methods. In seletected cases, following a good response to treatment for relapse (salvage therapy), high-dose chemotherapy followed by an autologous stem cell transplantation may also be an option.
In addition, new therapeutic approaches are being investigated. These include targeted drugs such as so-called multi-tyrosine kinase inhibitors, which can inhibit specific signalling pathways in tumour cells. Cabozantinib, a multi-tyrosine kinase inhibitor, has been investigated in clinical trials in patients with recurrent Ewing sarcoma and may be considered as a treatment option in selected situations. Other multi-targeted tyrosine kinase inhibitors, such as regorafenib, have also demonstrated efficacy in clinical trials.
When therapy with the goal of a cure is not an option any longer, maintenance of the patient’s life quality is of major importance. These include, for example, pain relief, the treatment of distressing symptoms and the maintenance of physical functions (palliative therapy).
Good to know: Clinical trials are constantly seeking to further improve treatment options and the prospects of recovery for these patients as well, for example by developing and testing new agents and new therapeutic approaches. Apart from tyrosine kinase inhibitors, other innovative treatment approaches are currently being researched. These include immunotherapies [see immuntherapy, such as antibody or cell therapies targeting tumour surface structures, drugs that target the genetic alterations characteristic of Ewing sarcoma, and substances that specifically interfere with DNA repair or cell division in tumour cells. Many of these approaches are still in the clinical trial phase, but offer hope for additional treatment options in the future.
Therapy optimising trials and registries
In the large paediatriac treatment centres, children and adolescents with Ewing sarcoma receive therapy according to standardised treatment plans (protocols). These protocols are designed by experts and aim at steadily improving the patients’ survival rates while also reducing the risk of therapy-related late effects. Therapy according to such treatment protocols is usually carried out within therapy optimising trials or registries.
Therapy optimising trials are standardised and controlled clinical trials that aim at steadily developing and improving treatment concepts for sick patients based on the current scientific knowledge. Patients who cannot participate in any study, for example because none is available or open for them at that time or since they do not meet the required inclusion criteria, respectively, are often included in a so-called registry. One goal of a registry is to collect treatment data for research questions. Furthermore, the registry centre supports the doctors at site with (non-commital) treatment recommendations based on the most recent data on best treatment options, in order to provide the patient with optimal therapy even without the framework of a clinical study.
The following treatment optimising trials and registries for patients with Ewing sarcoma are currently available in Germany (with international participation):
- Trial iEuroEwing: an international, multicentre, randomised therapy optimising trial (phase III trial) for patients with Ewing sarcoma (aged betwenn 2 and 49 years); patients with both localised disease (standard risk, SR) and metastatic disease (high risk, HR) are eligible for the trial, which was opened in 2022. The study examines, among other things, whether additional maintenance therapy can improve the prognosis for standard- and high-risk patients. The study coordination centre for Germany is located at the Children’s Hospital of the University of Essen under the direction of Prof. Dr. med. Uta Dirksen. Numerous treatment centres in and outside Europe are taking part in the study.
- iEwing-Registry: Since 2020, children and adolescents (as well as adults) with first diagnosis of Ewing sarcoma in Germany, who may not be treated in the context of any trial, can be registered with the International Euro Ewing Registry. The study coordination centre for Germany is located at the Children’s Hospital of the University of Essen (principal investigator is Prof. Dr. med. Uta Dirksen).
Note on ongoing phase I/II trials
As part of an international phase I trial (CESS-GD2), a study is being conducted in newly diagnosed patients (aged 12 months and over) with GD2-positive Ewing sarcoma to investigate whether the use of the therapeutic antibody dinutuximab beta [see monoclonal antibodies] in addition to standard chemotherapy (VDC/IE) could be considered as a future treatment option to improve the prognosis, particularly for high-risk patients.
Further phase I/II trials are being conducted with the aim of improving the currently poor prognosis for patients with recurrent and/or refractory Ewing sarcoma. One example is the PerVision Phase I/II trial. In the framework of this trial, children, adolescents and young adults with metastatic, fusion-gene-driven sarcomas receive a vaccination with patient-specific and tumour-specific peptides (small proteins) following completion of standard therapy. Another example is the phase I/II VolViln trial, which is investigating a new maintenance therapy for patients with Ewing sarcoma. As these therapies are still undergoing clinical trials, they can currently only be offered as part of relevant trials and subject to availability.
Aside from participating in this study, patients with metastasised or recurrent disease can also register with the INFORM-Registry. The registry systematically records genetic changes in tumours with the goal to be able to offer individually tailored therapies, thereby improving outcomes for relapse patients. The abbreviation “INFORM” stands for INdividualised Therapy FOr Relapsed Malignancies in Childhood. The study coordination centre is located at the Hopp-Childhood tumour centre in Heidelberg (KiTZ) under supervision of Prof. Dr. med. Olaf Witt.
Important note: The phase I/II trials mentioned above are merely examples. Please check with your healthcare team to find out which trials are currently open and which ones you or your child might be eligible for.
Prognosis
The chances of survival (prognosis) for children and teenagers with Ewing sarcoma depend on various factors. In particular, the extent of the tumour at diagnosis, its response to preoperative chemotherapy as well as its site and size are of major prognostic importance.
During the last decades, prognosis of children and teenagers with Ewing sarcoma has significantly been increased, thanks to modern diagnostic procedures and standardised combination therapies in the framework of therapy optimising trials.
While the probability of survival after radiation therapy alone was about 10 % in the 1960s, an average of more than 80 % of patients with local disease, meaning without visible metastases [see metastasis], can achieve long-term cure by a combination of local and chemotherapy today. Favourable prognosis usually requires complete tumour removal and good response to the chemotherapy that precedes surgery.
Patients with metastasised disease at primary diagnosis still have an unfavour-able prognosis despite of intensive chemotherapy (5-year survival rates are an average of about 20 %). Prognosis is more favourable for patients with single lung metastases that can be surgically removed, when compared to patients with bone or bone marrow metastases. Prognosis for patients with recurrent disease is similarly unfavourable. Most unfavourable is the prognosis for patients who develop early metastases after intensive frontline treatment. Currently active and future trials should help to improve the outcome of those patients as well.
Note: The survival rates mentioned in the text above are statistical values. Therefore, they only provide information on the total cohort of patients with osteosarcoma. They do not predict individual outcomes. In the context of cancer, the term "cure“ should rather be referred to as "free of cancer“, because current treatment regimens may help to destroy the tumour, but they are also frequently associated with numerous late-effects. Early detection and appropriate management of these long-term secondary effects typically require intensive rehabilitation and thorough long-term follow-up care, although a patient may have been "cured“ from the cancer.
 Brief information on Ewing Sarcoma (499KB)
Brief information on Ewing Sarcoma (499KB)References 
- Heesen P, Feuerriegel S, Ciobanu-Caraus O, Ranft A, Bhadri V, Brichard B, Collaud S, Cyprova S, Eich H, Ek T, Gelderblom H, Hardes J, Haveman L, Hartmann W, Hauser P, Jürgens H, Kanerva J, Kühne T, Raciborska A, Rascon J, Streitbürger A, Timmermann B, Uhlenbruch Y, Fuchs B, Dirksen U: Treatment effect heterogeneity of radiotherapy in localized Ewing sarcoma: A secondary analysis of the EURO-E. W.I. N.G. 99 and Ewing 2008 trial. European journal of cancer (Oxford, England : 1990) 2026 May 22; 243: 116825 [PMID: 42218103]
- Hubbard AK, Neyret-Kahn H, Müller-Nurasiyd M, Löw D, Strauch K, Lee OW, Raduski AR, Yang T, Zhou W, Stratton EW, Jay O, Grossetête S, Song AJ, Dutta D, Hutchinson AA, Hicks BD, Manning M, Liu J, Boyce C, Hartmann W, Dirksen U, Kulozik AE, Metzler M, Krumbholz M, Teumer A, Völzke H, Völker U, Schiffman JD, Khan J, Hudson MM, Ness KK, Wang Z, Janeway KA, Lupo PJ, Spector LG, Huang WY, Moore SC, Chanock SJ, Grünewald TGP, Delattre O, Machiela MJ: Genome-wide association study meta-analysis identifies susceptibility loci informing Ewing sarcoma etiology and potential mechanisms of risk. medRxiv 2026, [PMID: 41728338]
- Ronckers CM, Spix C, Grabow D, Erdmann F: German Childhood Cancer Registry - Annual Report 2022 (1980-2021). Institute of Medical Biostatistics, Epidemiology and Informatics (IMBEI) at the University Medical Center of the Johannes Gutenberg University Mainz 2025 [URI: https://www.kinderkrebsregister.de/ fileadmin/ kliniken/ dkkr/ pdf/ jb/ jb2022/ JB_2022_final.pdf]
- Kersting J, Ranft A, Bhadri V, Brichard B, Collaud S, Cyprová S, Eich H, Ek T, Gelderblom H, Hardes J, Haveman L, Hartmann W, Hauser P, Heesen P, Jürgens H, Kanerva J, Kühne T, Raciborska A, Rascon J, Rechl V, Streitbürger A, Timmermann B, Uhlenbruch Y, Dirksen U: Effect of Radiotherapy Dose on Outcome in Nonmetastatic Ewing Sarcoma. Advances in radiation oncology 2023, 8: 101269 [PMID: 37334316]
- Anderson WJ, Doyle LA: Updates from the 2020 World Health Organization Classification of Soft Tissue and Bone Tumours. Histopathology 2021, 78: 644 [PMID: 33438273]
- Zöllner SK, Amatruda JF, Bauer S, Collaud S, de Álava E, DuBois SG, Hardes J, Hartmann W, Kovar H, Metzler M, Shulman DS, Streitbürger A, Timmermann B, Toretsky JA, Uhlenbruch Y, Vieth V, Grünewald TGP, Dirksen U: Ewing Sarcoma-Diagnosis, Treatment, Clinical Challenges and Future Perspectives. Journal of clinical medicine 2021 Apr 14; 10 [PMID: 33919988]
- Gerrand C, Bate J, Seddon B, Dirksen U, Randall RL, van de Sande M, O'Donnell P, Tuckett J, Peake D, Jeys L, Saifuddin A, Grainger M, Whelan J: Seeking international consensus on approaches to primary tumour treatment in Ewing sarcoma. Clinical sarcoma research 2020, 10: 21 [PMID: 33292535]
- WHO Classification of Tumours Editorial Board: Soft Tissue and Bone Tumours. 5th ed. Lyon: IARC Press 2020 [URI: https://publications.iarc.who.int/ Book-And-Report-Series/ Who-Classification-Of-Tumours/ Soft-Tissue-And-Bone-Tumours-2020]
- Bauer S, Dirksen U, Schildhaus HU: [Systemic therapy of sarcomas : New biomarkers and therapeutic strategies]. Der Pathologe 2019, 40: 436 [PMID: 31243550]
- Dirksen U, Brennan B, Le Deley MC, Cozic N, van den Berg H, Bhadri V, Brichard B, Claude L, Craft A, Amler S, Gaspar N, Gelderblom H, Goldsby R, Gorlick R, Grier HE, Guinbretiere JM, Hauser P, Hjorth L, Janeway K, Jürgens H, Judson I, Krailo M, Kruseova J, Kuehne T, Ladenstein R, Lervat C, Lessnick SL, Lewis I, Linassier C, Marec-Berard P, Marina N, Morland B, Pacquement H, Paulussen M, Randall RL, Ranft A, Le Teuff G, Wheatley K, Whelan J, Womer R, Oberlin O, Hawkins DS, Euro-EWING 99 and Ewing 2008 Investigators: High-Dose Chemotherapy Compared With Standard Chemotherapy and Lung Radiation in Ewing Sarcoma With Pulmonary Metastases: Results of the European Ewing Tumour Working Initiative of National Groups, 99 Trial and EWING 2008. Journal of clinical oncology 2019,JCO1900915 [PMID: 31553693]
- Whelan J, Le Deley MC, Dirksen U, Le Teuff G, Brennan B, Gaspar N, Hawkins DS, Amler S, Bauer S, Bielack S, Blay JY, Burdach S, Castex MP, Dilloo D, Eggert A, Gelderblom H, Gentet JC, Hartmann W, Hassenpflug WA, Hjorth L, Jimenez M, Klingebiel T, Kontny U, Kruseova J, Ladenstein R, Laurence V, Lervat C, Marec-Berard P, Marreaud S, Michon J, Morland B, Paulussen M, Ranft A, Reichardt P, van den Berg H, Wheatley K, Judson I, Lewis I, Craft A, Jürgens H, Oberlin O, Euro-EWING99 and EWING-2008 Investigators: High-Dose Chemotherapy and Blood Autologous Stem-Cell Rescue Compared With Standard Chemotherapy in Localized High-Risk Ewing Sarcoma: Results of Euro-E. W.I. N.G. 99 and Ewing-2008. J Clin Oncol 2018 [PMID: 30188789]
- Worch J, Ranft A, DuBois SG, Paulussen M, Jürgens H, Dirksen U: Age dependency of primary tumor sites and metastases in patients with Ewing sarcoma. Pediatric blood & cancer 2018, 65:e27251 [PMID: 29856530]
- Whelan J, Hackshaw A, McTiernan A, Grimer R, Spooner D, Bate J, Ranft A, Paulussen M, Jürgens H, Craft A, Lewis I: Survival is influenced by approaches to local treatment of Ewing sarcoma within an international randomised controlled trial: analysis of EICESS-92. Clinical sarcoma research 2018, 8: 6 [PMID: 29610659]
- Dirksen U, Jürgens H: Ewing-Sarkom. in: Niemeyer C, Eggert A (Hrsg.): Pädiatrische Hämatologie und Onkologie, Springer-Verlag GmbH Deutschland, 2. vollständig überarbeitete Auflage 2018, 497 [ISBN: 978-3-662-43685-1]
- Gruenewald TGP, Cidre-Aranaz F, Surdez D, Tomazou EM, de Álava E,Kovar H, Sorensen PH, Delattre O, Dirksen U: Ewing sarcoma. Nature reviews. Disease primers 2018 Jul 5; 4: 5 [PMID: 29977059]
- Pappo AS, Dirksen U: Rhabdomyosarcoma, Ewing Sarcoma, and Other Round Cell Sarcomas. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2018 Jan 10; 36: 168 [PMID: 29220292]
- Gaspar N, Hawkins DS, Dirksen U, Lewis IJ, Ferrari S, Le Deley MC, Kovar H, Grimer R, Whelan J, Claude L, Delattre O, Paulussen M, Picci P, Sundby Hall K, van den Berg H, Ladenstein R, Michon J, Hjorth L, Judson I, Luksch R, Bernstein ML, Marec-Bérard P, Brennan B, Craft AW, Womer RB, Jürgens H, Oberlin O: Ewing Sarcoma: Current Management and Future Approaches Through Collaboration. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2015 Sep 20; 33: 3036 [PMID: 26304893]
- Haeusler J, Ranft A, Boelling T, Gosheger G, Braun-Munzinger G, Vieth V, Burdach S, van den Berg H, Jürgens H, Dirksen U: The value of local treatment in patients with primary, disseminated, multifocal Ewing sarcoma (PDMES). Cancer 2010, 116: 443 [PMID: 19924786]
- Burkhardt B, Reiter A, Landmann E, Lang P, Lassay L, Dickerhoff R, Lakomek M, Henze G, von Stackelberg A: Poor outcome for children and adolescents with progressive disease or relapse of lymphoblastic lymphoma: a report from the berlin-frankfurt-muenster group. Journal of clinical oncology 2009, 27: 3363 [PMID: 19433688]
- Gerth HU, Juergens KU, Dirksen U, Gerss J, Schober O, Franzius C: Significant benefit of multimodal imaging: PET/CT compared with PET alone in staging and follow-up of patients with Ewing tumors. Journal of nuclear medicine : official publication, Society of Nuclear Medicine 2007, 48: 1932 [PMID: 18006618]
- Wagner LM, McAllister N, Goldsby RE, Rausen AR, McNall-Knapp RY, McCarville MB, Albritton K: Temozolomide and intravenous irinotecan for treatment of advanced Ewing sarcoma. Pediatric blood & cancer 2007, 48: 132 [PMID: 16317751]
- Hawkins DS, Schuetze SM, Butrynski JE, Rajendran JG, Vernon CB, Conrad EU 3rd, Eary JF: [18F]Fluorodeoxyglucose positron emission tomography predicts outcome for Ewing sarcoma family of tumors. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2005, 23: 8828 [PMID: 16314643]