Retinoblastoma (brief information)
Retinoblastoma is a rare form of eye cancer that almost only occurs during childhood. The following pages provide information on possible causes and symptoms as well as on courses of the disease and its management, long-term follow-up and prognosis.
Author: Maria Yiallouros, Editor: Maria Yiallouros, Reviewer: Prof. Dr. med. Petra Ketteler, PD Dr. med. Eva Biewald, English Translation: Dr. med. Gesche Riabowol (nee Tallen), Last modification: 2026/02/05 https://kinderkrebsinfo.de/doi/e168130
Table of contents
General disease information
Retinoblastoma is a rare eye cancer arising from the retina, the inner layer of the eye, which is specialised in colour and light perception. It almost only occurs during childhood. There is a hereditary (congenital) and a non-hereditary form of the disease. Patients with the hereditary form have an inherited predisposition for the development of this cancer, while in the second case, the cancer develops spontaneously, meaning, it is initiated by a new genetic alteration in a retina cell.
Retinoblastoma may affect one or both eyes. In the majority of patients (about 60 %), only one eye is affected (unilateral retinoblastoma); in approximately 40 % of the children, the tumour develops in both eyes (bilateral retinoblastoma). Bilateral disease is almost always seen with hereditary retinoblastoma, while most unilateral retinoblastomas are non-hereditary. Tumours may develop either at one site in the eye (unifocal) or at multiple sites (multifocal). About a third of the affected children present with bilateral, multifocal tumours at initial diagnosis, or they develop additional tumours while the disease is progressing.
In general, retinoblastomas grow fast. They can spread within the eyeball, into the orbit, and along the optic nerve into the central nervous system (CNS). In advanced stages, they may also spread into other organs via the blood and/or lymphatic system. If not treated appropriately, the outcome will be lethal. Only in very few patients (1 %), the tumour disappears without treatment (spontaneous regression).
Incidence
Retinoblastoma is the most common childhood cancer of the eye. According to the German Childhood Cancer Registry, Mainz (Germany), about 45 children under 18 years of age (corresponding to approximately 4 out of 1.000.000) are diagnosed with this type of malignancy in Germany each year. This means that there is one child with retinoblastoma among 18,000 children born alive. Overall, however, retinoblastoma is a rare disease; it accounts for approximately 2 % of all malignant diseases in children and adolescents.
Most retinoblastomas are diagnosed in infants and small children, i.e. before the age of five. Usually, children with a bilateral retinoblastoma are younger than children with unilateral disease. Beyond the sixth year of life retinoblastomas are extremely rare. The average age of disease is about 1 year, with boys being slightly more affected than girls (gender ratio: 1.1 : 1).
Causes
The causes for a retinoblastoma to arise in the eye include certain genetic alterations (mutations) in the precursor cells of the retina. Such alterations may occur spontaneously in single retina cells before or after the child’s birth. Sometimes, however, the mutation may be present within the patient’s germ cells (and, thus, in all body cells), meaning, the disease is hereditary and can be passed on to the offspring, too.
In more than 50 % of patients, the altered gene is only found in the tumour itself. They have the non-hereditary (sporadic) type of retinoblastoma. Almost 50 % of retinoblastoma, though, are congenital (hereditary), which means, the affected children have a predisposition for the tumour to develop. About one quarter of these children (10 to 15 % of all patients) are presenting with a family story of retinoblastoma, this means, other family members are known to have this type of cancer, too (familial retinoblastom). In the remaining three quarters of patients with congenital retinoblastoma, the underlying genetical alterations are believed to have happened spontaneously.
Regardless of hereditary or non-hereditary, the mutations causing retinoblastoma are found in the retinoblastoma (RB1) gene, which is located on chromosome 13. Since in human cells chromosomes occur in pairs, there are also two retinoblastoma gene variants (so-called alleles) in each cell. Only when both retinoblastoma alleles are altered, the tumour will form. Since in hereditary retinoblastoma all retina cells initially contain one altered retinoblastoma gene variant already, “only” one additional mutation is required to induce tumour development. This is why the hereditary form presents at an early age, affects both eyes at (frequently) multiple sites (multifocal). Children who have inherited the altered gene have an almost 100% risk to develop retinoblastoma. Since there is a predisposition for developing additional cancers, hereditary retinoblastoma is also known as a cancer predisposition syndrome.
Symptoms
Very small retinoblastomas usually do not cause any complaints; the disease frequently progresses without any symptoms for a long period of time. Health problems usually occur with tumour growth into adjacent eye tissue. This can result in an impairment of vision or even loss of sight.

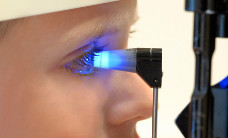
Child with leukocoria ("amourotic cat's eye")
© by courtesy of Prof. Dr. med. P. Ketteler
The most common and typical sign, which is found in about two thirds of patients, is a white flashing of the pupil (white pupillary reflex, leukocoria) under certain light conditions, for example, after a flash photograph was taken – in contrast to the well-known red or black appearance of the pupils in healthy eyes. This condition, also referred to as “amaurotic cat's eye” is caused by expansive tumour growth behind the lens. In addition, impairment of the visual axis may lead to crossed eyes (strabismus), depending on the source in up to almost 50% of patients. Less often, patients also complain about irritated, swollen, red or painful eye as a result of increased eye pressure caused by the growing tumour.
Warning signs indicative of retinoblastoma may be:
- “amaurotic cat’s eye” (leukocoria)
- crossed eyes (strabismus)
- impairment of vision or loss of sight / visual disturbances
- different colour of each eye (due to impaired pigmentation of the iris in the affected eye)
- irritated eye (redness of the white part of the eye, bulging of the eye, eye pain, without infection)
- visual impairment (involuntary eye movement, nystagmus)
- no shrinking of the pupil when exposed to bright light (mydriasis)
Occurrence of one or more of these symptoms, however, does not necessarily prove underlying retinoblastoma or any other malignancy. Some of these symptoms may be caused by relatively harmless health conditions that are not associated with cancer at all. Nevertheless, it is recommended to consider these symptoms as warning signs and therefore to consult a physician promptly upon their appearance. If retinoblastoma (or another malignant disease) is present, early diagnosis is the best precondition for a successful treatment of the disease.
Good to know in case of known, hereditary predisposition: children from families with a hereditary risk should – as long as they are known or assumed to have inherited the disease – have regular eye exams, so that retinoblastoma can be diagnosed and appropriate management initiated at an early stage (also see chapter "Diagnosis“).
Diagnosis
If a child’s medical history (anamnesis), presenting complaints and symptoms, as well as the physical examination, particularly the eye exam, are suggestive of retinoblastoma, the patient should immediately be referred to a children’s cancer centre. There, diagnostics and treatment can be initiated and performed by specialists with the necessary expertise in childhood cancer. Close collaboration between various specialists (such as paediatric oncologists, eye doctors, surgeons, radiologists, radiation oncologists, to name a few) as well as special tests are required to confirm the diagnosis and to determine the type of retinoblastoma (hereditary or non-hereditary) and how the disease has spread. Knowing these details is absolutely essential for optimal treatment planning and prognosis.
Examination of the eye
The most important initial diagnostic procedure for a young patient presenting with suspected retinoblastoma is the examination of the eye (fundoscopy). This includes looking at the fundus of both eyes with an ophthalmoscope and bright light. In case of retinoblastoma, fundoscopy also helps to assess the extent of the disease, which serves as a basis for staging. This considers the number, size and site of tumours as well as the potential extent beyond the retina or within the glass body of the eye. The examination is performed under anaesthesia with the pupil dilated to its maximum (called mydriasis) and serves to classify the disease into one of five disease groups (A-E according to the ICRB classification, see chapter "Treatment planning").
Imaging diagnostics and further tests
In order to assess the exact tumour extent (staging), fundoscopy is followed by diagnostic imaging such as ultrasound (sonography) and magnetic resonance imaging (MRI). Ultrasound serves to measure the tumour, for example. Using MRI of the eye-socket (orbit) and the brain helps to assess whether the disease is limited to the eye (known as intraocular retinoblastoma) or has spread into the eye layers, the optic nerve and/or the brain (extraocular retinoblastoma). Also, “trilateral” retinoblastoma (retinoblastoma plus brain tumour) can be diagnosed this way. For MRI, the patients are usually sedated. Also, a patient with first diagnosis of retinoblastoma should be seen by a paediatric oncologist.
Rarely, for example in patients with very advanced stages of the disease and/or prior to chemotherapy, additional tests may be performed, such as chest X-ray examination, spinal tab (lumbar puncture), bone marrow puncture, and/or a MRI of the spine or the whole body. The latter also serves to find out whether the bones are involved and, thus, has meanwhile replaced skeletal scintigraphy (bone scan).
Once all necessary diagnostics are completed, the doctors will discuss the best treatment options with you.
Genetic testing
Since hereditary retinoblastoma cannot be ruled out in any patient, human genetics are consulted at primary diagnosis as well, and, following informed consent, the patient’s blood is analysed for molecular genetics (genetic testing). This includes a DNA analysis to identify genetic alterations (mutations) that are typical for retinoblastoma. Finding those mutations implicates that the child has hereditary retinoblastoma. In this case, the diagnostic procedures are not limited to the affected child only. Genetic testing and eye exams are also required for the patient’s siblings and parents in order to clarify the risk of the disease within the family and hence to be able to initiate appropriate early detection and monitoring measures, since it is possible that the siblings have inherited the abnormal gene as well.
Good to know: genetic consulting and testing are crucial parts of the care of children and families with retinoblastoma.
Early detection of known hereditary disease
As of today, children in families with retinoblastoma (familial retinoblastoma) can be tested for the defective gene immediately after birth (the probability of having inherited it is 50 %). Precondition is, that the mutation that is harboured in the family has been identified. This is not always possible, since the alterations within the retinoblastoma gene can be very heterogeneous. In case the child has inherited the mutations (or if this is uncertain), an eye exam is strongly recommended in order to detect and treat tumours as early as possible. Close monitoring should be maintained as long as the immature retina cells, which tend to degenerate, have fully differentiated, which means until about the child’s fifth year of life. Affected individuals will also be followed-up regularly until adulthood.
Treatment planning
After the diagnosis has been confirmed, therapy is planned. In order to design a highly individual, risk-adapted treatment regimen for the patient, certain individual factors influencing the patient’s prognosis (called risk factors or prognostic factors) are being considered before and during treatment (risk-adapted treatment strategy).
Extent of retinoblastoma at the time of diagnosis is an important prognostic factor and hence a crucial criterion regarding the choice of the most feasible treatment. Also, unilateral versus bilateral disease is being considered. Knowing the extent of the disease helps the physicians to predict how the patient’s disease might respond to a certain form of treatment, whether there is remaining vision of one or both eyes to be expected after therapy and how high the risk of progressive or recurrent disease is. Also, the patient’s age and clinical condition as well as the knowledge about a genetic predisposition for the disease are integrated in treatment planning.
Stages of retinoblastoma
Based on the extent, retinoblastoma is divided into different stages (also known as classification). Consideration is primarily focussed on whether the retinoblastoma is intraocular, thereby affecting the eye(s) only, or if the disease has spread to other parts of the body (extraocular retinoblastoma). Tumour extent is always assessed separately for both eyes.
For staging of intra- and extraocular retinoblastoma, various classification systems have been developed:
- Classification of intraocular retinoblastoma is mostly based on the „International Classification of Retinoblastoma“ (ICRB, Philadelphia version). The ICRB staging system (also called ABC classification) considers size and site of the tumour as well as the existence and extent of spread within the vitreous body. This results in a classification of retinoblastoma into 5 stages (A to E).
- Extraocular retinoblastoma is classified according to the International Re-tinoblastoma Staging System (IRSS). This considers in particular whether the tumour has infiltrated the choroid, the dermis and/or the optic nerve, or whether it has spread into the orbit, the central nervous system (meningeosis) and/or even has distant metastases. Depending on the diagnosis, four stages are differentiated: IRSS I-IV. A retinoblastoma that won’t be enucleated is assigned to stage 0.
Another classification, the TNM classification for retinoblastoma, merges information on intra- and extraocular disease.
For further details on the classification of intra- and extraocular retinoblastoma according to ICRB and ISSR, please see our information on „Stages of retinoblastoma (classification)“.
Treatment
Children with retinoblastoma should be taken care of in a children’s cancer centre by specialists, because they provide the expertise necessary to manage this rare type of childhood malignancy.
Current treatment options for children with retinoblastoma include surgery, chemotherapy (local and systemic), radiotherapy (brachytherapy or percutaneous radiotherapy), laser therapy, cryotherapy and thermotherapy. For some patients with an advanced disease, high-dose chemotherapy followed by autologous stem cell transplantation may be an option, too.
The appropriate type of treatment is individually assessed for each patient with retinoblastoma. It is generally based on whether one or two eyes are affected by the tumour, how far the disease has already spread (intraocular or extraocular retinoblas-toma and exact stage of disease) and whether visual acuity can be expected for one or both eyes after treatment. The child’s age at diagnosis is important for therapy planning, as well. The major aim of treatment is to completely destroy or remove the tumour and to cure the disease. In this context, survival has fundamental priority over preserving visual acuity.
In general, the following two treatment strategies currently exist:
- saving the eye by using radio-, laser-, cryo-, thermo, and/or chemotherapy
- surgical removal of the tumour by removal of the eyeball (enucleation) and sometimes also additional (adjuvant) forms of therapy for maintenance of the treatment success
Eye-preserving therapy is, whenever an option, preferred. For single, small retinoblastomas, a so-called ophthalmological local (focal) therapy is the gold standard. If tumours are already too large for this approach, chemotherapy for reducing tumour size may be an option (chemoreduction) in order to make subsequent local therapy feasible. The eye-preserving strategy aims at inactivating the tumour while preserving as much vision as possible, but also making sure that the treatment is efficient enough so the patient won’t die from the disease.
If retinoblastoma has been diagnosed at an advanced stage, enucleation, i.e. the removal of the eyeball, is usually necessary. If the tumour has spread into other parts of the body (metastasised), treatment may also include chemotherapy and/or radiation therapy in addition to surgery. Some situations even require a high-dose chemotherapy followed by autologous stem cell transplantation to be performed.
The various therapy options will be explained below.
Eye-preserving therapy options (intraocular retinoblastoma)
Eye-preserving therapies include the ophthalmological focal therapies on the one hand and chemotherapy (systemic and/or local) on the other hand. Percutaneous radiotherapy (radiation from outside the body through the skin) is rarely indicated in case of intraocular retinoblastoma.
Ophthalmological local treatment options (focal therapy)
With the help of ophthalmological local therapies, smaller intraocular tumours (ICRB stages A and partially B) can be treated successfully in an eye-preserving manner. Possible treatment options are laser therapy (also called laser coagulation or photocoagulation), cryotherapy (or cryocoagulation), thermotherapy and local radiation therapy (brachytherapy). The individual treatment choice is based, above all, on the tumour’s size and localisation:
- Laser therapy is particularly used for smaller tumours (smaller than 2 mm in height); it includes guiding a laser beam through the pupil onto the tumour, while the patient is under general anaesthesia. The tumour is being destroyed by the heat of the laser beam.
- Cryotherapy uses extremely low temperatures. The tumour is detected by using an ophthalmoscope and a metal probe and then frozen multiple times. The cold-sensitive tumour cells are thereby destroyed. This approach is feasible for tumours (up to 3-4 mm in height) that are located in the front part (periphery) of the retina. Local spread into the vitreous body can be treated this way as well.
- Thermotherapy (or thermochemotherapy) is a form of laser therapy that is combined with a systemically applied chemotherapy. It is particularly used for tumours in the back of the eye pole.
- Brachytherapy (short distance radiation) is used for single retinoblastomas (between 4-6 mm in height) located at easily accessible sites. It includes the surgical placement of a radioactive applicator (such as a Ruthenium applicator) onto the sclera in the area of the tumour and leaving it there until the desired radiation dose has been given (usually a few days). Radiation is targeting the tumour only. Therefore, a high total radiation dose can be applied to the tumour while protecting adjacent tissue. Over weeks, months or even years, the radiation-sensitive retinoblastoma is transformed into inactive scar tissue.
All these treatment approaches may be used both alone and in combination with other therapies. They may also be feasible for consolidation in patients with retinoblastomas that could be reduced in size by systemic chemotherapy.
Chemotherapy
Chemotherapy uses drugs (so-called cytostatics) that can kill fast-dividing cells, such as cancer cells, or inhibit their growth, respectively.
Systemic chemotherapy for intraocular retinoblastoma (chemoreduction)
For retinoblastoma patients, systemic chemotherapy – a chemotherapy that can control tumour cells everywhere in the body (systemically) - is often applied in addition to other therapy methods to increase their efficiency or to consolidate treatment success. Usually, a combination of various agents is used (polychemotherapy). They are given via a vein (intravenous chemotherapy), from where they distribute in the whole body via the blood stream, thus also reaching the tumour’s blood vessels.
While, in the past, systemic chemotherapy has primarily been used in advanced disease stages (tumour spread beyond the orbit) or following enucleation, it plays an important role today also for retinoblastomas with an extent that is limited to the eye (intraocular retinoblastoma). The goal of chemotherapy in the context of eye-preserving therapy is to reduce the size of existing tumours in the eye (so-called chemoreduction), so that they can subsequently be further controlled by ophthalmological local treatment forms. This approach is meant to avoid percutaneous radiotherapy as well as enucleation in as many patients as possible.
Local chemotherapy
In order to accumulate high concentrations of chemotherapy directly within the eye while also minimizing or avoiding the risk of adverse events of intravenous (systemic) chemotherapy (which typically affects the whole body), local chemotherapy has been used for eye-preserving therapy for some time, too. The necessity of percutaneous radiotherapy can be minimized by these relatively new treatment options as well.
- For intra-arterial chemotherapy, a cytostatic (for example melphalan) is given directly into an artery of the eyeball. This is done by the following procedure: After the child has been put to sleep by anaesthesia, the doctor inserts a catheter into a big artery in the child’s groin and – monitored by X-ray examination – pushes it carefully through the con-nected arteries and past the heart all the way up into the region of the artery of the eye which is to be treated. From there, the substance distributes to the downstream blood vessels including those located in the retinoblastoma. Intra-arterial chemotherapy is very effective for eye-preserving therapy. It may be used during frontline treatment as well as following a preceeding therapy.
- Intravitreal chemotherapy (IViC) is a therapy option for patients with retinoblastoma cells within the vitreous body (vitreous body spread). By giving melphalan or certain other agents (such as topotecan), vitreous body spread can usually be well-controlled and hence the eye preserved.
Surgical removal of the eye (enucleation)
For a long time, the surgical removal of the affected eye (enucleation) was the most frequently applied and efficient treatment option for retinoblastoma patients and the only possibility for complete tumour removal and, thus, cure of the disease.
Still, enucleation is the therapy of choice for advanced intraocular disease (ICRB stage E, partly also stage D). Hence, it is indicated for patients who present with a retinoblastoma that, due to its size, does not allow local therapy and has already caused severe loss of vision at diagnosis, with no remaining preservable eye sight to be expected by eye-preserving treatment options. This is frequently the case in one-sided (unilateral) retinoblastoma, which are usually already advanced at the time of diagnosis. In bilateral retinoblastomas, tumours often have grown differently, and hence, both eyes present with different stages of the disease. In those cases, often the more affected eye is being removed, as long as both eyes cannot be preserved.
Sometimes, if in case of bilateral retinoblastoma systemic chemotherapy is being considered a treatment option for the less affected eye, enucleation of the more affected eye may be postponed to await the results of that chemotherapy. For, in some cases, the chemotherapy aiming especially at treating the less affected eye may as well cause a significant tumour regression in the more affected eye, allowing eye-preserving therapy strategies instead of enucleation after all. However, there is no alternative to enucleation in case the worse eye is already blind or there is infiltration of the anterior segment of the eye or the optic nerve.
For tumour removal, the complete eyeball needs to be removed along with a rather long part of the optic nerve. Patients who underwent complete surgical removal of the tumour usually do not require any additional treatment after surgery (adjuvant therapy). However, if histology of the removed eye reveals that the tumour has surpassed certain boundaries (extraocular retinoblastoma) or – due to various risk factors – bears an elevated risk for the development of metastases, respectively, further treatment (so-called adjuvant therapy) may be necessary (see chapter “Treatment of patients with extraocular retinoblastoma” below).
Treatment of patients with extraocular retinoblastoma
Patients who, following enucleation, present with certain histological risk factors (choroid coat-infiltration, involvement of the sclera or the optic nerve), thereby revealing tumour spread outside the eye, require additional (adjuvant) therapy in order to reduce the risk of further tumour spread via the bloodstream into distant regions of the body and/or via the optic nerve into the brain. Even in case metastases already exist, additional treatment is required. The more advanced the disease, the more intense and complex the treatment. Treatment options are: systemic chemotherapy alone, combined chemotherapy and radiation therapy (the latter locally limited to the orbit) as well as a multimodal therapy including chemotherapy, high-dose chemotherapy with autologous stem cell transplantation.
Trials and registries
Since retinoblastoma is a very rare disease (around 47 children are diagnosed in Germany per year), so far only few data have been acquired that can be used as a basis for treatment strategies which both consider a patient’s individual risk of recurrent disease (risk-adapted therapy) and are statistically proven (evidence-based therapy).
In contrast to treatments of other childhood cancers, a standardised therapy plan (protocol) for the management of retinoblastoma still needs to be established, for example as a therapy optimising trial. Therefore, the Retinoblastoma Registry (RB Registry) was opened for patients in Germany and Austria by the end of 2013. This clinical registry aims at gathering various data on retinoblastoma over several years, such as incidences and clinical courses of the different types of the disease as well as their response to different treatment forms, in order to fill the information gap mentioned above and, thus, to optimise current therapy and outcomes.
All children and adolescents under the age of 18 years who have been diagnosed with retinoblastoma or have a germline mutation of the retinoblastoma (RB1) gene and who have not yet received any retinoblastoma-specific treatment can be included in the RB-Registry. The headquarters of the RB-Registry are located in the Childhood Cancer Centre of the University of Essen, Germany. The registry is coordinated by Prof. Dr. Petra Ketteler.
For patients with localised, unilateral retinoblastoma who, due to risk factors, require enucleation as part of their initial treatment, the Europe-wide clinical trial EURbG2 will be launched in 2026, initially in France and subsequently in other European countries such as Germany and Austria.
Prognosis
Due to continuously optimised diagnostic procedures and treatment forms, more than 95 % of children with retinoblastoma can be cured of their disease today. Children with unilateral retinoblastoma still have one completely unaffected eye with normal vision. Their life quality may not differ from that of their healthy peers at all. Also, most patients with retinoblastoma in both eyes will keep a certain amount of visual acuity in at least one eye.
The individual prognosis primarily depends on the patient's stage of disease at diagnosis as well as on whether they have the hereditary or non-hereditary type of retinoblastoma.
Patients with tumour growth limited to the eye(s) only (intraocular retinoblastoma) usually have a bigger chance of successful treatment and, thus, a more favourable prognosis than children whose disease is further progressed at diagnosis.
The overall prognosis of children with hereditary retinoblastoma is generally less favourable compared to the outcome of patients with the non-hereditary form, because, regardless of the treatment, hereditary retinoblastoma is associated with a higher risk of developing a second malignant tumour somewhere else in the body (for example a soft tissue sarcoma or an osteosarcoma). This risk increases if radiation therapy forms part of the treatment. About 5 % of children with hereditary, unilateral retinoblastoma develop the disease in the contralateral eye within one and a half years after diagnosis of the first tumour.
 PDF Brief Information on Retinoblastoma (313KB)
PDF Brief Information on Retinoblastoma (313KB)References 
- Ronckers CM, Spix C, Grabow D, Erdmann F: German Childhood Cancer Registry - Annual Report 2022 (1980-2021). Institute of Medical Biostatistics, Epidemiology and Informatics (IMBEI) at the University Medical Center of the Johannes Gutenberg University Mainz 2025 [URI: https://www.kinderkrebsregister.de/ fileadmin/ kliniken/ dkkr/ pdf/ jb/ jb2022/ JB_2022_final.pdf]
- Dunkel IJ, Piao J, Chantada GL, Banerjee A, Abouelnaga S, Buchsbaum JC, Merchant TE, Granger MM, Jubran RF, Weinstein JL, Saguilig L, Abramson DH, Krailo MD, Rodriguez-Galindo C, Chintagumpala MM: Intensive Multimodality Therapy for Extraocular Retinoblastoma: A Children's Oncology Group Trial (ARET0321). Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2022 Jul 12;:JCO2102337 [PMID: 35820112]
- Stanulla M, Erdmann F, Kratz CP: Risikofaktoren für Krebserkrankungen im Kindes- und Jugendalter. Monatsschrift Kinderheilkunde 169, 30-38 2021 [DOI: 10.1007/s00112-020-01083-8]
- Reschke M, Biewald E, Bronstein L, Brecht IB, Dittner-Moormann S, Driever F, Ebinger M, Fleischhack G, Grabow D, Geismar D, Göricke S, Guberina M, Le Guin CHD, Kiefer T, Kratz CP, Metz K, Müller B, Ryl T, Schlamann M, Schlüter S, Schönberger S, Schulte JH, Sirin S, Süsskind D, Timmermann B, Ting S, Wackernagel W, Wieland R, Zenker M, Zeschnigk M, Reinhardt D, Eggert A, Ritter-Sovinz P, Lohmann DR, Bornfeld N, Bechrakis N, Ketteler P: Eye Tumors in Childhood as First Sign of Tumor Predisposition Syndromes: Insights from an Observational Study Conducted in Germany and Austria. Cancers 2021 Apr 14; 13 [PMID: 33919815]
- Kiefer T, Schlüter S, Bechrakis NE, Bornfeld N, Göricke S, Ketteler P, Ting S, Geismar D, Biewald E: Intraarterial Chemotherapy for Retinoblastoma - Initial Experiences of a German Reference Centre. Klinische Monatsblatter fur Augenheilkunde 2021, 238: 788 [PMID: 34376009]
- Ketteler P, Hülsenbeck I, Frank M, Schmidt B, Jöckel KH, Lohmann DR: The impact of RB1 genotype on incidence of second tumours in heritable retinoblastoma. European journal of cancer (Oxford, England : 1990) 2020, 133: 47 [PMID: 32434110]
- Bornfeld N, Biewald E, Bauer S, Temming P, Lohmann D, Zeschnigk M: The Interdisciplinary Diagnosis and Treatment of Intraocular Tumors. Deutsches Arzteblatt international 2018 Feb 16; 115: 106 [PMID: 29510820]
- Temming P, Eggert A: Retinoblastom, in: Niemeyer C, Eggert A (Hrsg.): Pädiatrische Hämatologie und Onkologie. Springer-Verlag GmbH Deutschland 2018 [ISBN: 978-3-662-43686-8]
- Ritter-Sovinz P, Temming P, Wackernagel W, Tarmann L, Langmann G, Benesch M, Lackner H, Karastaneva A, Schwinger W, Seidel M, Sperl D, Strenger V, Sorantin E, Urban C: Retinoblastom - Klinische Symptome, Diagnostik und Management. Monatsschrift Kinderheilkunde 165, 764-771 2017 [DOI: 10.1007/s00112-017-0364-3]
- Temming P, Arendt M, Viehmann A, Eisele L, Le Guin CH, Schündeln MM, Biewald E, Astrahantseff K, Wieland R, Bornfeld N, Sauerwein W, Eggert A, Jöckel KH, Lohmann DR: Incidence of second cancers after radiotherapy and systemic chemotherapy in heritable retinoblastoma survivors: A report from the German reference center. Pediatric blood & cancer 2017, 64: 71 [PMID: 27567086]
- Temming P, Viehmann A, Arendt M, Eisele L, Spix C, Bornfeld N, Sauerwein W, Jöckel KH, Lohmann DR: Pediatric second primary malignancies after retinoblastoma treatment. Pediatric blood & cancer 2015, 62: 1799 [PMID: 25970657]
- Temming P, Eggert A, Bornfeld N, Sauerwein W, Göricke S, Lohmann DR: [Diagnosis and treatment of retinoblastoma: current strategies for effective tumour control and preservation of vision]. Klinische Monatsblatter fur Augenheilkunde 2013, 230: 232 [PMID: 23508752]
- Shields CL, Shields JA: Intra-arterial chemotherapy for retinoblastoma: the beginning of a long journey. Clinical & experimental ophthalmology 2010, 38: 638 [PMID: 20584015]
- Shields CL, Shields JA: Retinoblastoma management: advances in enucleation, intravenous chemoreduction, and intra-arterial chemotherapy. Current opinion in ophthalmology 2010, 21: 203 [PMID: 20224400]
- Shields CL, Mashayekhi A, Au AK, Czyz C, Leahey A, Meadows AT, Shields JA: The International Classification of Retinoblastoma predicts chemoreduction success. Ophthalmology 2006, 113: 2276 [PMID: 16996605]
- Chantada G, Doz F, Antoneli CB, Grundy R, Clare Stannard FF, Dunkel IJ, Grabowski E, Leal-Leal C, Rodríguez-Galindo C, Schvartzman E, Popovic MB, Kremens B, Meadows AT, Zucker JM: A proposal for an international retinoblastoma staging system. Pediatric blood & cancer 2006, 47: 801 [PMID: 16358310]
- Shields CL, Shields JA: Basic understanding of current classification and management of retinoblastoma. Current opinion in ophthalmology 2006, 17: 228 [PMID: 16794434]