Neuroblastoma – Brief information
Neuroblastoma is a malignancy of the sympathetic nervous system, which particularly occurs in early childhood. This text provides information about the characteristics of this disease, its frequencies, causes, symptoms, diagnosis, treatment, and prognosis.
Author: Maria Yiallouros, Editor: Maria Yiallouros, Reviewer: Prof. Dr. med. Frank Berthold, Prof. Dr. med. Thorsten Simon, Prof. Dr. med. Angelika Eggert, English Translation: Dr. med. Gesche Riabowol (nee Tallen), Last modification: 2026/06/18 https://dx.doi.org/10.1591/poh.neurobl.patinfo.kurz.1.20120611
Table of contents
General information on the disease
Neuroblastomas are malignant solid tumours. They arise from degenerate immature cells of the sympathetic nervous system, which is part of the autonomic nervous system.
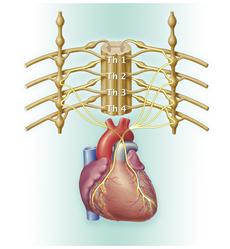
Neuroblastomas can occur at any site of sympathetic nervous tissue. They most frequently develop in the medulla of the adrenal gland (adrenal medulla, about 50 %) and within the nervous plexus on both sides of the spinal column, the so-called sympathetic trunk. If the sympathetic trunc is affected, neuroblastoma can occur at any spinal level, such as in the abdominal, pelvic, chest or neck region. In most cases (for about 75 %), the tumour can be found in the abdomen, about one fifth of tumours in the chest or neck region.
Some neuroblastomas are limited to their original site, others spread into close lymph nodes. At initial diagnosis, about half of the patients present with tumour spread (metastases) to the bone marrow, bones, distant lymph nodes or liver, less frequently with brain, lung or skin metastases. A typical feature of biologically favourable neuroblastomas is their ability to spontaneously regress (see chapter „Courses of disease“).
Incidence
Neuroblastoma accounts for approximately 5.3 % of all malignant diseases in childhood and adolescence, thereby representing one of the most frequent solid tumour type in this age group following tumours of the central nervous system (CNS tumours, brain tumours). According to the German Childhood Cancer Registry (Mainz), about 120 children and adolescents under 18 years of age are newly diagnosed with a Neuroblastoma each year. Hence, 11 out of 1,000,000 children and adolescents between 0 and 17 years are diagnosed with this disease.
Since Neuroblastoma are embryonal tumours, they are most frequent in early childhood: 90 % of all affected patients are younger than 6 years old; about 46 % are newborns and babies in their first year of live. The average age of the patients at diagnosis is 15 months, with boys being (by 40 %) more affected than girls (gender ratio: 1.3:1). Nevertheless, older children, adolescents and, seldomly, adults may also be affected.
Causes
The underlying causes for the development of neuroblastoma are still not completely understood. It is known that the disease is initiated by a malignant transformation (degenerate) of immature cells of the sympathetic nervous system. The impaired development of not yet completely matured (embryonal) nerve cells most certainly starts before birth and can be a consequence of chromosomal aberrations and/or genetic changes (mutations).
Various genetic changes have been identified in neuroblastoma cells so far. However, these are very heterogenous, meaning that there is no specific genetic transformation that can be consistently observed in every tumour. Overall, a series of genetic and also epigenetic modifications may be responsible for the development of neuroblastoma. Hereditary transmission of the disease has, for most patients, not been proven yet.
However, families with increased incidences of neuroblastoma and related tumours over multiple generations have been described. About 1–2 % of patients have such a family history, and they often present with more than one primary tumour. Neuroblastoma can also be associated with so-called cancer predisposition syndromes, congenital diseases characterized by mutations that (compared to healthy individuals) are connected with a higher risk of developing a malignancy at a younger age. Cancer predisposition syndromes playing a role in the development of neuroblastoma are, for example, Hirschsprung`s disease or the Undine syndrome.
In most patients, however, the disease arises as a result of spontaneous mutations or other genomic changes in the cells’ DNA. Whether external factors (such as environmental factors, parental exposure to stress factors at work, certain drugs, nicotin or alcohol abuse during pregnancy) play a role as well has not been elucidated yet.
Symptoms
Many patients with neuroblastoma do not complain about any health problems (symptoms). They are rather randomly diagnosed with the tumour, for example during a routine check-up or an ultrasound or X-ray examination that is being done for a different reason. Symptoms usually occur at advanced stages of tumour growth, when the tumour has spread (metastasis) or impairs adjacent tissue.
In addition, symptoms can vary dependent on the site of the tumour or the metastases [see metastasis]. Palpable tumours or metastases may be first symptoms, some children present with an abdominal or cervical swelling. Tumours of the abdomen or the adrenal gland can be associated with rather unspecific symptoms such as abdominal pain, constipation, bloating or diarrhea; compression of the ureter may lead to urine retention. Tumours in the chest can compress the lungs, thereby causing cough, pneumonia or shortness of breath. Neuroblastomas that are close to the spinal column (tumours of the sympathetic trunk) can grow into the spinal canal and subsequently result in neurological symptoms such as pain, impaired micturition or bowel incontinence.
High blood pressure (hypertension) as well as persistent diarrhea are, in rare cases, caused by the hormonal activity of the tumour. Tumours in the neck or upper chest region can result in the so-called Horner`s syndrome. This is defined by by one eye presenting with a very small pupil, a drooping eyelid and with the eyeball displaced backwards (also known as unilateral miosis, ptosis and enophthalmus). Further changes of the eyes may include small bruises of the eyelid (eyelid ecchymosis) as well as, in cases of advanced disease, bruises around one or both eyes (so-called monocular haematoma). A rare variant of the disease is neuroblastoma with a so-called opsoclonus-myoclonus-ataxia syndrome (OMAS).
Bone metastases – which particularly occur in the long bones of arms and legs as well as in the skull and the bones around the eyes – can result in bone pain. Some patients with bone pain present with a limp. In case of severe bone marrow involvement, patients may present with low red blood cell counts (anaemia) low platelet counts (thrombocytopenia) and low white blood cell counts (leukopenia) and, subsequently, an increased risk of infection and bleeding.
General symptoms that are suspicious of, mostly advanced stage of, neuro-blastoma are:
- fatigue, listlessness, decreased performance levels, frailty, pallor
- persistent fever with unknown cause, sweats
- tumours or swelling in the abdominal or neck region; lymph node swellings
- distended abdomen
- constipation or diarrhea, abdominal colics
- loss of appetite, nausea, vomiting and subsequent weight loss
- bone pain
- bruises around the eyes
Good to know: the occurrence of one or more of these symptoms does not necessarily mean that a person has neuroblastoma. Many of these symptoms can have rather harmless causes. Nevertheless, any of these complaints should be reported to a doctor in order to find out what causes them.
Diagnosis
If the doctor thinks that the young patient’s medical history (anamnesis) and physical examination are suspicious of a neuroblastoma, he will refer the child immediately to a hospital with a childhood cancer program (paediatric oncology clinic), where further diagnostics can be initiated and performed by childhood cancer experts. Close collaboration between various specialists (such as paediatric oncologists, paediatric neurosurgeons, paediatric radiologists, to name a few) is required, both to find out whether the patient indeed suffers from a neuroblastoma and, if so, to determine the tumour type and the extent of the disease. Knowing these details is absolutely essential for optimal treatment planning and prognosis.
Laboratory tests
Aside from comprehensive medical history taking and physical exam, laboratory tests play a major role in confirming the diagnosis. Most patients with neuroblastoma present with elevated levels of certain bodily substances in the blood or urine, which are used as “tumour markers” to confirm the diagnosis (and, in particular, to monitor the treatment efficacy during the course of the disease). Relevant tumour markers for neuroblastoma are certain catecholamines or their metabolites, respectively (vanillinic acid, homovanillinic acid), as well as the neuron-specific enolase (NSE).
Diagnostic imaging
Additional tests to establish the diagnosis and to rule out other diseases (such as Wilms tumour, phaeochromocytoma) include diagnostic imaging: ultrasound is usually sufficient to determine both site and size of most abdominal or cervical neuroblastomas. X-rays help to check on chest and lungs.
In order to identify very small tumours and their impact on adjacent structures, magnetic resonance imaging (MRI) with and without contrast agent is used. Sometimes, computed tomography (CT) is applied instead. In general, however, MRI is the preferred imaging technique, since it does not involve ionising radiation but magnetic fields, thereby not causing radiation exposure.
Tests to assess tumour spread
To assess or, respectively, rule out metastases [see metastasis] and also to determine additional features of the primary tumour, total body scintigraphy is performed using the low radioactive agent 123-Iodo-metaiodobenzylguanidine (short 123I-MIBG scintigraphy or MIBG scan). This substance accumulates in catecholamine-producing tissues and thus in almost all neuroblastomas (with the exception of so-called MIBG-negative neuroblastomas). If the MIBG scan is negative, i.e. shows no abnormalities (as MIBG has not been taken up), other scintigraphic techniques can serve as alternatives, such as a positron emission tomography (PET) with radioactively labelled glucose (18F-fluoride-deoxyglucose, short FDG). Both techniques are combined with CT or MRI, respectively.
Since scintigraphy is not sufficient to identify very mild bone marrow involvement, obtaining a bone marrow sample is required for all patients. Bone marrow is obtained via bone marrow puncture or bone marrow punch biopsy from at least two different sites, while the patient is sedated or under short anaesthesia, and subsequently analysed under the microscope using techniques that are specific for looking at cancer cells. Patients with metastases also undergo an MRI of the brain to rule out central nervous system involvement. A total body MRI may be indicated for patients with advanced disease in order to identify potential bone metastases.
Obtaining a tumour sample (biopsy)
In general, final diagnosis can be established only by microscopic (histological) analysis of tumour tissue. A tumour sample is usually obtained by surgery. Molecular genetic analyses [see molecular genetics] allow conclusions regarding the grade of the tumour’s malignancy. In fact, certain genetic changes (mutations) in the tumour DNA (such as the so-called MYCN amplification or 1p deletion) as well as the existence of certain genetic variants (experts call it unfavourable genetic signature) correlate with unfavourable prognosis, while lack of these changes or other mutations can be associated with a favorable outcome.
Additional genetic changes were identified in neuroblastoma cells a few years ago (for example changes of the ALK gene or the so-called telomerase activation), which can be used therapeutically in case of relapse as long as they were identified in the initial tumour. ALK alterations can also be used therapeutically in case of relapse, provided they have been detected in the patient’s initial tumour.
Tests before treatment begins
Depending on the type of treatment being considered, further tests are needed in order to assess the condition of different organs. These include, especially prior to chemotherapy, an examination of the heart (electrocardiography, echocardiography), a hearing test (for example, audiometrym BERA hearing test and/or otoacoustic emissions), special diagnostics for determining kidney function (kidney ultrasound, kidney nuclear medicine scan) as well as an X-ray examination of the hand to determine the child’s bone age. In order to recognize certain treatment-related adverse reactions and subsequently manage them appropriately and as early as possible, many of these tests are regularly being repeated during the course of treatment and their results compared to those obtained by the initial tests.
Good to know: Not all of the above-mentioned tests will be done for every single patient. On the other hand, additional tests not mentioned here may be required individually. Ask the doctor which diagnostics are necessary and why.
Treatment planning
After the diagnosis has been confirmed, therapy is planned. In order to design an individual, risk-adapted treatment regimen for the patient, certain factors influencing the patient’s chance of recovery (prognosis) – called risk factors or prognostic factors – are being considered during treatment planning (risk-adapted treatment strategy).
The stage of the disease at the time of diagnosis is of major importance and, thus, a very significant prognostic factor. Aspects like the tumour’s extent and the extent of its surgical removal are included in the staging of a neuroblastoma (see table below). Additional relevant prognostic factors include the patient’s age as well as the histological and, particularly, the molecular genetic characteristics of the tumour, which reveal information on its growth and spread pattern (see chapter "Diagnosis“). All these factors are included in treatment planning in order to achieve the best outcome possible for each patient.
More information on the stages of neuroblastoma can be found in the following paragraphs.
Stages of neuroblastoma
The extent of the tumour within the body has usually a great impact on the chance of cure and is, therefore, an important criterion for choosing the feasible treatment strategy. The classification of neuroblastoma according to the stages of the disease considers the tumour size, lymph node involvement and existence of metastases [see metastasis]. Additional criteria that play a role in staging depend on the applied classification system. Two classification systems are currently in use: the INSS and the INRG classification.
- INSS Classification: following the traditional, international staging system, which has been used in Germany for a long time (International Neuroblastoma Staging System, INSS), the extent of surgery has been added to the factors mentioned above; hence, the exact assessment of the stage was only possible after surgery.
- INRG Classification: while the INSS classification is still being applied, the International Neuroblastoma Risk Group Staging System (INRG) is now the internationally valid classification system. The INGR system evaluates the stage of the disease prior to surgery based on established risk factors, which have been identified by diagnostic imaging (MRI or CT). Such a risk factor (called Image Defined Risk Factor, short: IDRF) is, for example, a walling-in of large blood vessels by the tumour. Aside from the surgical options, the patient’s age, and the molecular characteristics of the tumour, the histology of neuroblastoma is also included in the staging.
Both classification systems are presented by the following tables. The INSS-classification differentiates between the localized stages 1-3, the metastatic stage 4 and the metastatic infant neuroblastoma stage 4S. The INGR staging differentiates the localized stages L1 and L2 under consideration of certain risk factors as well as the advanced (metastatic) stages M and (see table below).
|
Tumour stages according to INSS |
Description |
Tumour stages ac-cording to INRG |
Description |
|---|---|---|---|
|
1 |
Localized, completely resected tumour |
L1 |
Localized tumour not involving vital structures (as defined by the list of IDRFs) and confined to one body compartment |
|
2a |
Localized, incompletely resected tumour Involvement of only one side of the spinal column No lymph node involvement in the tumour region |
||
|
2b |
Localized, completely or incompletely resected tumour Involvement of only one side of the spinal column Involvement of adjacent lymph nodes at the same side |
L2 |
Localized tumour with presence of one or more IDRFs |
|
3 |
Incompletely resected tumour with extension to the other side of the spinal column or Involvement of lymph nodes on the contralateral side |
||
|
4 |
Distant metastases (for example in bone marrow, bone, liver, skin, distant lymph nodes and other organs) |
M |
Distant metastatic disease (except stage MS tumour) |
|
4S |
Only occurring in infants (0-18 months according to current criteria); metastases only in skin, liver and/or, minimally, in the bone marrow |
MS |
Metastatic disease in children younger than 18 months, with metastases confined to skin, liver, and/or bone marrow |
With the exception of patients with stage 4S or MS, respectively, patients with less advanced disease usually have a more favorable prognosis than patients with advanced stages (such as stages 3 and 4). Patients with less favorable stages usually require a more intensive therapy than patients with more favourable prognosis (see chapter "Course of treatment“).
Courses of the disease
The courses of a neuroblastoma disease vary individually, particularly depending on the growth pattern of the tumour and its stage at initial diagnosis. Hence, neuroblastoma can be localized at diagnosis but also can have infiltrated other tissue and lymph nodes, either close or distant (see paragraph “Stages of neuroblastoma” above). A special feature of the biologically and clinically more favourable neuroblastomas is the fact that they can spontaneously turn into benign tumours (so-called differentiation) or spontaneously recede (spontaneous regression).
Tumour growth and spread
Particularly in children older than 18 months, neuroblastomas frequently grow fast and uncontrolled and spread within the whole body mostly via the bloodstream, but sometimes also via the lymphatic system. Metastases preferably develop in the bone marrow (in 90 % of the patients), bone (60 %), distant lymph nodes (20 %) and liver (17 %), less frequently in the brain (9 %), skin (2 %) and lungs (1 %). These cases are considered as stage 4 or M, respectively.
Tumour maturation (differentiation)
Neuroblastomas with a favourable biological profile may undergo tumour maturation (known as differentiation), either spontaneously or as a result of a mild chemotherapy. In this case, the tumours do not disappear during observation and may require further surgery later on. However, the prognosis for these patients remains very favourable. Even high-risk neuroblastomas with unfavourable biology can some-times mature under chemotherapy. However, these patients have a high risk of recurrence, so the planned intensive high-risk therapy must still be completed.
In addition, there are patients who, at the time of diagnosis, already have a fully matured neuroblastical tumour, known as a ganglioneuroma. These patients are generally older. Since ganglioneuromas do not respond to chemotherapy, they must always be removed surgically. The risk of recurrence is minimal, even following incomplete tumour removal.
Tumour regression (spontaneous regression)
A high number of neuroblastomas regress spontaneously (tumour regression). The tumour cells die by a form of self-initiated cell death, a process that scientists call apoptosis. The spontaneous tumour regression is particularly and almost regularly observed in neuroblastomas that are diagnosed in infancy or early childhood. This can affect localised tumours and even metastatic neuroblastomas.
A special case is stage 4S or MS in young patients. In these patients, in addition to the tumour itself, the liver is often enlarged due to extensive metastasis. These initially diagnosed metastases may still rapidly grow in the beginning, thereby pushing on abdominal organs and lungs and reaching a life-threatening extent, so that mild chemotherapy is required. Subsequently, however, they may regress.
However, spontaneous tumour regression does not only occur in stage 4S (MS) in infants, but are also observed in older children with localized neuroblastoma stage 1 to 3 or L1 to L2, respectively.
Information on the different stages of the disease can be found in the paragraph “Treatment planning” (see above).
Treatment
Treatment of children and adolescents with neuroblastoma should take place in a children's hospital with a paediatric oncology program. Only such a childhood cancer centre provides highly experienced and qualified staff (doctors, nurses, psychosocial support service), since they are specialised and focussed on the diagnostics and treatment of children and teenagers with cancer according to the most advanced treatment concepts. The doctors in these centres collaborate closely with each other. Together, they treat their patients according to treatment plans (protocols) that are continuously optimised.
The goal of treatment is to eliminate the cancer while keeping the risk of side effects and late sequelae as low as possible.
Treatment methods
Therapy of a neuroblastoma patient is based on the individual situation of the disease and the probability of relapse. For some patients, surgery to remove the tumour or to obtain a tumour sample can be sufficient, while others require the combination of multiple treatment methods in order to improve the chances of cure.
The treatment methods described in the following paragraphs represent the current standard of treatment in Germany. New treatment concepts are continuously being evaluated in the framework of clinical studies, so the treatment may sometimes differ from this standard in single patients.
Treatment of neuroblastoma includes surgery, chemotherapy and radiotherapy. In addition, patients with a high risk of relapse usually undergo high-dose chemotherapy with subsequent autologous stem cell transplantation as well as immunotherapy with antibodies. Further treatment methods, such as the MIBG-therapy, a treatment with radioactively labelled metaiodobenzylguanidine, may be indicated in these patients as well.
The goal of surgery is to remove the tumour and/or to gain a tissue sample. Chemotherapy uses drugs (so-called cytostatics) that can kill fast-dividing cells, such as cancer cells, or inhibit their growth, respectively. Since one cytostatic agent alone may not be capable of destroying all tumour cells, a combination of cytostatics that function in different ways is given (polychemotherapy). The goal is to eliminate as many malignant cells as possible. A high-dose chemotherapy is even more intense: it is capable of eliminating resistant neuroblastoma cells. Since the high doses of cytostatics given according to this treatment strategy also lead to the destruction of the blood-forming cells in the bone marrow, the patient will receive blood stem cells in a second step. These stem cells are obtained from the patient’s blood or bone marrow prior to high-dose chemotherapy and are given back right after this treatment (so-called autologous stem cell transplantation, SCT). Radiotherapy (radiation therapy), which is required in high-risk patients, is done using energy-rich, electromagnetic radiation, given through the skin to the tumour region. Radiation causes DNA damage in tumour cells, thereby leading to cell death.
The individual treatment choice is based on the tumour`s extent at diagnosis and after surgery as well as on is growth characteristics (tumour biology) and the patient’s age. Taken together, the more advanced the tumour stage and the higher the risk of an aggressive tumour growth or a relapse after the end of treatment, the more intensive therapy will be.
Since treatment of neuroblastoma can be associated with side effects, supportive treatment measures (supportive therapy) are also applied to prevent and/or treat these side effects. Here you will find further information on supportive care as well as recommendations for home, the latter of which may be helpful during or after chemo- and radiotherapy.
Course of treatment
At the beginning of treatment, every patient is being assigned to a certain risk or treatment group. The current treatment guidelines consider three different groups of therapy: low-risk group, intermediate-risk group and high-risk group. Each of these treatment groups is associated with a different treatment plan. This strategy allows a risk-adapted therapy that is individually adjusted to the patient.
Treatment within the low-risk group (watch-and-wait group)
The low-risk group (watch-and-wait group) includes patients, who, due to localized tumour growth and/or their age, are not put at risk by the watch-and-wait approach of this treatment strategy. The proof of lack of unfavourable molecular genetic tumour characteristics (such as the MYCN amplification or, in part, also the 1p deletion) is crucial for the assignment of a patient to this watch-and-wait group. Hence, the watch-and-weight group includes patients with one of the following disease features:
- Stage 1 (INSS), age 0-21 years, no MYCN amplification
- Stage 2 (INSS), age 0-21 years, neither MYCN amplification nor 1p deletion
- Stage 3 (INSS), age 0-2 years, neither MYCN amplification nor 1p deletion
- Stage 4S (INSS), age-range is limited, but according to INRG stage MS extended to 0-18 months, neither MYCN amplification nor 1p deletion
Course of treatment: Treatment for patients with low-risk neuroblastoma is usually limited to surgical tumour removal, provided this can be done safely. Otherwise, given the high rate of spontaneous tumour regression, even an incomplete operation or a tissue sample (biopsy) is sufficient.
If the patient is in good clinical condition, no chemotherapy or radiotherapy is given. However, the patient is monitored closely (by regular physical exams, ultrasound, magnetic resonance imaging and tumour markers). In the first year, patients undergo follow-up at least every six weeks, from the second to fifth year at least every three months, and after that at least every six to twelve months. The type of follow-up exam varies based on whether the patient has residual tumour and if the tumour is sufficiently assessable by ultrasound.
If the residual tumour starts to grow again during the watch-and-wait period, or if it progresses and/or starts causing symptoms that require treatment (such as reduced clinical status, nutrition problems, weight loss, hypertension, urine transport problems), usually a low-dose chemotherapy is indicated in order to initiate tumour regression. Treatment consists of up to four cycles of combination chemotherapy with doxorubicin, vincristine and cyclophosphamide. Alternatively, carboplatin and etoposide may be given as well.
Chemotherapy will be ended as soon as the tumour stops growing. If the tumour does not show a clear reduction in size as the condition progresses, further surgery may be required to remove the tumour or to reduce symptoms. The latter applies in particular to patients with stage 4S, whose tumour has the potential to significantly grow before it regresses.
Treatment within the intermediate-risk group
The intermediate-risk group includes patients with a more advanced stage of the disease and/or older patients as well as patients with certain unfavourable molecular genetic characteristics (1p deletion). A MYCN amplification needs to be ruled out. Patients with the following disease constellations are assigned to the intermediate-risk group:
- Stage 2 (INSS), age 0-21 years, with deletion in chromosome 1p, but no MYCN amplification
- Stage 3 (INSS), age 0-21 years, with deletion in chromosome 1p, but no MYCN amplification
- Stage 3 (INSS), age 2-21 years, neither MYCN amplification nor 1p deletion
- Stage 4 (INSS), age-range is limited, but according to INRG stage MS extended to 0-18 months, no MYCN amplification
Course of treatmment: treatment consists of surgery or, if not feasible, a biopsy first. After that, chemotherapy is given. It consists of six blocks of intensive chemotherapy (induction chemotherapy) and four blocks of a, slightly lower-dose, maintenance chemotherapy. In case only a biopsy could be performed prior to chemotherapy, surgical tumour removal is being strived for after the first cycles of induction chemotherapy because, frequently, the tumour shrinks during chemotherapy.
For induction treatment, the standard combination chemotherapy regimen contains doxorubicin, vincristine and cyclophosphamide or carboplatin, etoposide and vindesine, respectively, which are administered alternately and as intravenous infusions over hours or days. Maintenance therapy consists of cyclophosphamide, which is mostly given as pills or as a solution.
If active tumour tissue is still found after the intensive chemotherapy, children older than 18 months will receive radiation therapy (with a radiation dose of 36 to 40 gray) of the residual tumour during maintenance chemotherapy. An initially residual tumour (for example in cases with initial biopsy only) may sometimes shrink so much during chemotherapy that complete surgical removal during or after this treatment phase becomes feasible. Total duration of treatment is about one year.
Treatment within the high-risk group
Patients with stages 1, 2, 3 or 4S whose tumour harbours a MYCN amplification, as well as all patients with stage 4 who are older than 18 months are assigned to the high-risk group. The treatment regimen for patients with high risk neuroblastoma is quite extensive.
Course of treatment: following surgery or biopsy, patients receive chemotherapy with multiple agents for about five months (so-called induction chemotherapy). The current standard induction regimen consists of six blocks of chemotherapy with alternating combinations of cisplatin, etoposide and vindesine or vincristine, dacarbacine, ifosfamide and doxorubicin, respectively. In between or after those cycles, a second-look surgery is usually performed, preferably with complete tumour removal. After that, patients undergo high-dose chemotherapy followed by autologous stem cell transplantation (duration: about six weeks). For patients with MIBG-positive residual tumour, additional treatment with radioactively-labelled 123-Iodo-metaiodobenzylguanidine, (123I-MIBG therapy) is a feasible option. In these cases, the 123I -MIBG therapy is given prior to the high-dose chemotherapy.
Following high-dose treatment, radiation of the tumour bed with a dose of 21 gray (Gy) and an immunotherapy with the antibody dinutuximab beta is given. The aim of this treatment phase (also known as maintenance or post-consolidation therapy) is to eliminate potentially residual tumour cells. In case of an active residual tumour, a total radiation dose of up to 36 Gy is recommended. The total duration of therapy can last up to two years.
Note on trial HR-NBL2 for patients with high-risk neuroblastoma: As part of the study for high-risk patients, which has been open since 2023, patients are receiving a different induction chemotherapy regimen to the standard one (COJEC). For high-dose chemotherapy and radiotherapy, two different treatment regimens are being compared in each of the two treatment arms. Patients are randomised to the treatment arms if the respective treatment is eligible [see randomisation].
Trials and registries
In Germany, the majority of the children and adolescents with neuroblastoma receive therapy according to the treatment plans of clinical trials or registries. The aim of clinical trials is to gradually improve treatments under strictly controlled conditions, so that the risks to participants are kept to a minimum.
Regardless of whether patients take part in a clinical trial or not, they are all recorded in a so-called registry. Such a registry serves primarily to accompany the patients‘ therapy scientifically. Furthermore, the registry centre supports the doctors at site with (non-commital) treatment recommendations based on the most recent data on best treatment options, in order to provide the patient with optimal therapy even without the framework of a clinical trial.
Currently, the following therapy optimising trials and registries are available for patients with neuroblastoma in Germany:
- Trial HR-NBL2 (high-risk neuroblastoma trial 2.0): international, multicentre, randomisid ther-apy trial (phase III studies) for patients with high-risk neuroblastoma; the SI-OP Europe Neuroblastoma (SIOPEN) study has been open for patient recruit-ment in Germany since 2023. The German headquarters are located at the Department of Paediatric Oncology and Haematology at the Charité in Berlin (principal investigator: Prof. Dr. med. Hedwig E. Deubzer). The study analyses different treatment concepts of induction chemotherapy as well as high-dose chemotherapy and radiotherapy with the aim of further improving the prognosis of patients. Numerous paediatric oncology centres throughout Germany as well as in other European and non-European countries participate in this trial.
- NB Registry 2016: Registry for newborns, infants, children and adolescents (as well as adults with a newly diagnosed or recurrent neuroblastic tumour (neuroblastoma, gan-glioneuroblastoma, ganglioneuroma); the registry was opened in 2017 after the end of therapy trials NB2004 und NB 2004-HR. Its major aim is to gather knowledge about the incidence, disease courses and long-term sequelae, thereby improving prognosis. Decision-making on the treatment regimen is done by the attending physician with the support of the therapy recommenda-tions from the study centre. Registering for the registry does not exclude future recruitment for a clinical study. The registry centre is located at the University Children’s Hospital in Cologne with Prof. Dr. med. Thorsten Simon as acting head.
Note on trials for patients with relapse: For patients with relapsed or refractory high-risk neuroblastoma, several phase I/II studies do exist, on which further, up-to-date information can be obtained by contacting the study headquarters in Berlin, Cologne and Greifswald).
Prognosis
Appraising the probability of cure from neuroblastoma remains a challenge. Both the extent of the disease and the aggressiveness of the tumour play a role. Children with stage 4S neuroblastoma as well as most patients with localized disease (stages 1 and 2) have a very good prognosis with 10-year survival rates of partially more than 90 %. Also, younger children (under 18 months) with stage 3 tumours have a favourable prognosis, as long as they don’t present with unfavourable molecular tumour features. For older children with metastatic neuroblastoma (stage 4), cure rates are, with only 50 % or less, still unfavourable, despite the intensive therapy regimen.
Note: The above-mentioned numbers are statistical values. Therefore, they only provide information on the total cohort of patients with this type of tumour. They do not predict individual outcomes. Please ask the doctor, who is responsible for your child, for competent information on her individual prognosis.
Further information
The information provided in this chapter are primarily based on the references cited below, are considering the current guidelines and treatment protocols for children and adolescents with neuroblastoma and have been created in collaboration with the study centre for neuroblastoma. Please contact your attending physician in case of further questions.
 Brief information on Neuroblastoma (PDF) (359KB)
Brief information on Neuroblastoma (PDF) (359KB)Literature 
- Simon T, Thole T, Castelli S, Timmermann B, Jazmati D, Schwarz R, Fuchs J, Warmann S, Hubertus J, Schmidt M, Rogasch J, Körber F, Vokuhl C, Schäfer J, Schulte JH, Deubzer H, Rosswog C, Fischer M, Lang P, Langer T, Astrahantseff K, Lode H, Hero B, Eggert A: GPOH Guidelines for Diagnosis and First-line Treatment of Patients with Neuroblastic Tumors, update 2025. Klinische Padiatrie 2025, 237: 117 [PMID: 40345224]
- Ronckers CM, Spix C, Grabow D, Erdmann F: German Childhood Cancer Registry - Annual Report 2022 (1980-2021). Institute of Medical Biostatistics, Epidemiology and Informatics (IMBEI) at the University Medical Center of the Johannes Gutenberg University Mainz 2025 [URI: https://www.kinderkrebsregister.de/ fileadmin/ kliniken/ dkkr/ pdf/ jb/ jb2022/ JB_2022_final.pdf]
- Hero B, Schuster U, Weisser, K: Neuroblastom - Informationen für Eltern. Fördergesellschaft Kinderkrebs-Neuroblastom-Forschung e.V [URI: https://neuroblastoma.de/ ratgeber-fuer-betroffene-eltern/ ]
- Simon T: Leitlinie: Neuroblastom. S1-Leitlinie 025-008 (Leitlinie der Gesellschaft für Pädiatrische Onkologie und Hämatologie) AWMF-online 2019 [URI: https://register.awmf.org/ assets/ guidelines/ 025-008l_S1_Neuroblastom_2019-07-abgelaufen.pdf]
- Eggert A, Simon T, Hero B, Lode H, Ladenstein R, Fischer M, Berthold F: Neuroblastom. in: Niemeyer C, Eggert A (Hrsg.): Pädiatrische Hämatologie und Onkologie Springer Verlag GmbH GDeutschland 2006, 2018, 2. vollständig überarbeitete Auflage 2018, 420 [ISBN: 978-3-662-43685-1]
- Ackermann S, Cartolano M, Hero B, Welte A, Kahlert Y, Roderwieser A, Bartenhagen C, Walter E, Gecht J, Kerschke L, Volland R, Menon R, Heuckmann JM, Gartlgruber M, Hartlieb S, Henrich KO, Okonechnikov K, Altmüller J, Nürnberg P, Lefever S, de Wilde B, Sand F, Ikram F, Rosswog C, Fischer J, Theissen J, Hertwig F, Singhi AD, Simon T, Vogel W, Perner S, Krug B, Schmidt M, Rahmann S, Achter V, Lang U, Vokuhl C, Ortmann M, Büttner R, Eggert A, Speleman F, O'Sullivan RJ, Thomas RK, Berthold F, Vandesompele J, Schramm A, Westermann F, Schulte JH, Peifer M, Fischer M: A mechanistic classification of clinical phenotypes in neuroblastoma. Science (New York, N.Y.) 2018 Dec 7; 362: 1165 [PMID: 30523111]
- Handgretinger R, Matthes-Martin S, Lang P: Hämatopoetische Stammzelltransplantation. in: Niemeyer C, Eggert A (Hrsg.): Pädiatrische Hämatologie und Onkologie, Springer-Verlag GmbH Deutschland 2. vollständig überarbeitete Auflage 2018, 17 [ISBN: 978-3-662-43685-1]
- Berthold F, Spix C, Kaatsch P, Lampert F: Incidence, Survival, and Treatment of Localized and Metastatic Neuroblastoma in Germany 1979-2015. Paediatric drugs 2017, 19: 577 [PMID: 28786082]
- Fischer J, Pohl A, Volland R, Hero B, Dübbers M, Cernaianu G, Berthold F, von Schweinitz D, Simon T: Complete surgical resection improves outcome in INRG high-risk patients with localized neuroblastoma older than 18Â months. BMC cancer 2017 Aug 4; 17: 520 [PMID: 28778185]
- Oberthuer A, Berthold F, Hero B, Till H: Neuroblastome, in: Solide Tumoren im Kindesalter. Fuchs J (Hrsg.) Schattauer GmbH: Stuttgart 2012, 77 [ISBN: 978-3-7945-2786-1]
- Brisse HJ, McCarville MB, Granata C, Krug KB, Wootton-Gorges SL, Kanegawa K, Giammarile F, Schmidt M, Shulkin BL, Matthay KK, Lewington VJ, Sarnacki S, Hero B, Kaneko M, London WB, Pearson AD, Cohn SL, Monclair T, International Neuroblastoma Risk Group Project: Guidelines for imaging and staging of neuroblastic tumors: consensus report from the International Neuroblastoma Risk Group Project. Radiology 2011, 261: 243 [PMID: 21586679]
- Øra I, Eggert A: Progress in treatment and risk stratification of neuroblastoma: impact on future clinical and basic research. Seminars in cancer biology 2011, 21: 217 [PMID: 21798350]
- Maris JM: Recent advances in neuroblastoma. The New England journal of medicine 2010 Jun 10; 362: 2202 [PMID: 20558371]
- Monclair T, Brodeur GM, Ambros PF, Brisse HJ, Cecchetto G, Holmes K, Kaneko M, London WB, Matthay KK, Nuchtern JG, von Schweinitz D, Simon T, Cohn SL, Pearson AD, INRG Task Force: The International Neuroblastoma Risk Group (INRG) staging system: an INRG Task Force report. Journal of clinical oncology 2009, 27: 298 [PMID: 19047290]
- Cohn SL, Pearson AD, London WB, Monclair T, Ambros PF, Brodeur GM, Faldum A, Hero B, Iehara T, Machin D, Mosseri V, Simon T, Garaventa A, Castel V, Matthay KK, INRG Task Force: The International Neuroblastoma Risk Group (INRG) classification system: an INRG Task Force report. Journal of clinical oncology 2009, 27: 289 [PMID: 19047291]
- Oberthuer A, Theissen J, Westermann F, Hero B, Fischer M: Molecular characterization and classification of neuroblastoma. Future oncology (London, England) 2009, 5: 625 [PMID: 19519203]
- Fischer M, Spitz R, Oberthür A, Westermann F, Berthold F: Risk estimation of neuroblastoma patients using molecular markers. Klinische Padiatrie 2008, 220: 137 [PMID: 18478485]
- Hero B, Simon T, Spitz R, Ernestus K, Gnekow AK, Scheel-Walter HG, Schwabe D, Schilling FH, Benz-Bohm G, Berthold F: Localized infant neuroblastomas often show spontaneous regression: results of the prospective trials NB95-S and NB97. Journal of clinical oncology 2008, 26: 1504 [PMID: 18349403]