Pancreatic tumours (Brief information)
Tumours of the pancreas are rare tumours among children and adolescents. They can be benign or malignant. With this text, you will receive information on the disease, symptoms, causes, diagnosis, treatment and prognosis of malignant pancreatic tumours, such as pancreatoblastoma, solid pseudopapillary tumours (SPT) and carcinomas of the pancreas.
Author: Prof. Dr. med. Dominik T. Schneider, Dr. med. Ines Brecht, Editor: Maria Yiallouros, English Translation: Dr. med. Gesche Riabowol (nee Tallen), Last modification: 2024/05/31 https://kinderkrebsinfo.de/doi/e175364
Table of contents
Both benign and malignant tumours can develop in the pancreas. This, however, happens rarely in childhood and adolescence; in Germany, less than ten children or teenagers are diagnosed with such a tumour per year.
Hence, there are no therapy studies providing the optimal treatment strategy that has been proven for many patients, as there are for other tumours in childhood and adolescence. However, the experts of the Registry for Rare Diseases in Paediatrics (German abbreviation: STEP, for "Seltene Tumorerkrankungen in der Pädiatrie") have summarized their experiences as well as the experiences of international research groups on the following pages.
Since every child is different, it is important to individually adjust the treatment to every single patient. Therefore, “STEP” offers free advice via a tumourboard attended by multidisciplinary experts. Feel free to have your attending physician contact the experts at “STEP” at step@klinikumdo.de. Also, by registering your child with the „STEP“ registry, you get the option to help expanding the experience with these rare tumours, thereby helping other children who are diagnosed with such a disease in the future.
The following paragraphs provide information on the most frequent pancreatic tumours in childhood and adolescence.
Introduction to the pancreas
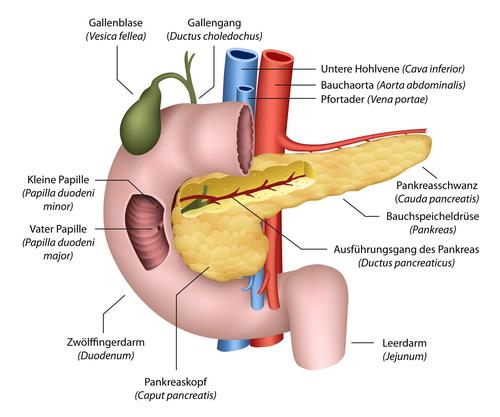
The pancreas is located behind the stomach at the level of the kidneys in the upper back of the abdomen. The longish organ has a head and a tail. The head is surrounded by the duodenum.
(© bilderzwerg - Fotolia.com)
The pancreas consists of both exocrine and endocrine gland tissue. The exocrine gland cells are responsible for the production of digestive juices (digestive enzymes), which are secreted into the duodenum via the pancreatic duct. The endocrine tissue contains cell groups that are scattered all over the organ (so-called Langerhans islets). These produce the hormones insulin and glucagon, which control blood sugar levels.
Pancreatoblastoma
Pancreatoblastoma is an embryonal, malignant tumour of the pancreas, which is very rare. Recently, a total of 63 published case reports over a period of 20 years (2000-2020) have been summarized in a review article on the incidence of this disease. The patients' mean age at diagnosis is five years of age, however, the tumour is also found in very small children as well as in adolescents and in adults.
Cause: how does pancreatoblastoma develop?
So far, the causes of pancreatoblastoma are still unknown. However, the tumour has been observed more frequently in children with certain genetic co-morbidities (such as Beckwith-Wiedemann syndrome). This implies that alterations within these genes may also play a role in the development of pancreatoblastoma.
Symptoms: what are the signs of the disease?
Since the pancreas is sort of hidden in the abdomen, symptoms are often unspecific and only appear with rather large tumours. Frequent symptoms are stomach aches and an increase in abdominal circumference or a palpable tumor in the abdomen, respectively. Weight loss and fatigue may also occur while jaundice is rather rare. The stool can be colourless, thereby presenting with a colour that reminds of clay or chalk.
Diagnosis: what kinds of tests are required?
Usually, the paediatrician will take a thorough history (anamnesis) and perform a physical examination followed by a routine blood test. This includes a complete blood count (CBC) as well as liver function tests and tests of the pancreatic enzymes. As a specific parameter, alpha-fetoprotein (AFP) levels are determined, since they can be elevated with these types of tumour. AFP also works as a marker for tumour response to therapy during treatment. Therefore, AFP is called a tumour marker.
Imaging of the tumour is also important to obtain information on its location, growth pattern and spread in adjacent or even distant organs and tissues. This is done by using different imaging techniques such as ultrasound (sonography), magnetic resonance tomography (MRT, layered scanning of the body using magnetic fields and radio waves) or computed tomography (CT, detailed x-ray slices of the body).
Therapy: how is treatment done?
The tumour should be surgically removed completely. If this is impossible because of vital organs and blood vessels being already involved, chemotherapy is used to shrink the tumour prior to surgery. For this, the agents doxorubicin and cisplatin are used. Treatment is based on the strategy applied to childhood liver tumours (hepatoblastomas), which are biologically similar. Radiation therapie (radiotherapy) is done only rarely, in case neither chemotherapy nor surgery appears to be a promising approach.
Important: the tumours behave aggressively and require optimal treatment adapted to the patient’s risk of relapse. Hence, please do not hesitate to seek advice.
Prognosis: what are the chances of cure?
The chances of cure (prognosis) for children and adolescents with pancreatoblastoma are slightly more favourable than for adults with malignant pancreatic tumours. It is assumed that more than half of the patients achieve long-term survival.
A major impact on prognosis is whether the tumour has already reached an advanced stage, meaning whether there is spread (metastasis) to the liver, lungs, bones or brain. Usually, the more extended the spread at initial diagnosis, the more unfavourable are the chances of cure.
Particularly important is an optimal treatment planning with the goal to achieve total gross tumour resection. Due to the rarity of these tumours and their complicated anatomical location, surgery should only be performed after discussion in an interdisciplinary tumour board and by experienced surgeons. In addition, patients and families require thorough professional care (by paediatricians and paediatric surgeons) during and after treatment.
Solid pseudopapillary tumours (SPT) of the pancreas
Solid pseudopapillary tumours (SPT) are rare tumours of the pancreas. Since 1959, more than 750 new diagnoses have been reported in the literature. Striking is the high incidence in young women. In about 10 % of all patients, children and adolescents are affected.
Cause: how do SPT develop?
So far, nothing is known about the mechanism of how solid pseudopapillary tumours develop. There are also no known risk factors.
Symptoms: what are the signs of the disease?
Symptoms can vary a lot. Most patients are completely free of complaints and the tumour is detected during a routine physical exam. Sometimes, however, pain or a palpable tumour in the upper abdomen, back pain, jaundice, nausea and/or vomiting are reported.
Diagnosis: what kinds of tests are required?
Usually, the paediatrician will take a thorough history (anamnesis) and perform a physical examination followed by a routine blood test. This includes a complete blood count (CBC) as well as liver function tests and tests of the pancreatic enzymes. Different from pancreatoblastoma (please see above), no specific lab value has been identified that could be used as a tumour marker. However, alpha-fetoprotein (AFP) levels should be determined to rule out a malignant pancreatoblastoma.
Imaging of the tumour is also important to obtain information on its location, growth pattern and spread in adjacent or even distant organs and tissues. This is done by using different imaging techniques such as ultrasound (sonography), magnetic resonance tomography (MRT, layered scanning of the body using magnetic fields and radio waves) or computed tomography (CT, detailed x-ray slices of the body).
Therapy: how is treatment done?
Goal of therapy is the total removal (gross total resection) of the tumour tissue. In children and adolescents this can be managed particularly well. If total resection is not an option, the children also benefit from partial removal. Tumour spread (metastasis) is rare and should be removed surgically as well. Chemo- and radiotherapy do not play a role for therapy and are only done in very rare scenarios. Do not hesitate to seek advice in complicated situations.
Prognosis: what are the chances of cure?
Patients with a solid pseudopapillary tumour usually have a very good prognosis. 97 % of the patients are still alive after five years. Within about 10 years after diagnosis, 15 % of the patients develop spread to the liver and peritoneum and, more rarely, also lymph node metastases [see metastasis]. Therefore, it is important that patients are being cared for by experienced, specially trained paediatricians even after the end of treatment.
Other pancreatic tumours in childhood and adolescence
So-called neuroendocrine tumours can arise from the hormone-producing cells of the pancreas. In childhood and adolescence, these tumours are found mostly in patients who are suffering from multiple endocrine neoplasia syndrome (MEN syndrome); the tumour is then caused by an inherited mutation of a cancer gene.
Neuroendocrine tumours can be benign or malignant; the latter are carcinomas, which can partially take an aggressive course and develop metastasis. In Germany, children and adolescents with these tumours are registered with the GPOH-MET Registry, which also considers other malignant childhood endocrine tumours. The registry provides advice upon optimal treatment, too.
Aside from these, there are very rare other childhood carcinomas of the pancreas, for example the acinar cell carcinoma or adenocarcinoma. Treatment of these tumours is mostly based on the recommendations for adult patients. As for prognosis, gross total resection is crucial for these tumours as well. Please don’t hesitate to seek advice.
 PDF Brief information on pancreatic tumours (187KB)
PDF Brief information on pancreatic tumours (187KB)
Author: Maria Yiallouros
Status: 14/10/2023, glossary included
References 
- Liu T, Zhao T, Shi C, Chen L: Pancreatoblastoma in children: Clinical management and literature review. Transl Oncol 2022, 18: 101359 [PMID: 35180620]
- Hippert F, Desing L, Diez S, Witowski A, Bernbeck B, Abele M, Seitz C, Erdmann F, Brecht I, Schneider DT: Rare Tumors in Children and Adolescents - the STEP Working Group's Evolution to a Prospective Registry. Klinische Padiatrie 2022, 234: 146 [PMID: 34798669]
- Achajew A, Brecht IB, Radespiel-Tröger M, Meyer M, Metzler M, Bremensdorfer C, Spix C, Erdmann F, Schneider DT, Abele M: Rare pediatric tumors in Germany - not as rare as expected: a study based on data from the Bavarian Cancer Registry and the German Childhood Cancer Registry. European journal of pediatrics 2022, 181: 2723 [PMID: 35478271]
- Bien E, Roganovic J, Krawczyk MA, Godzinski J, Orbach D, Cecchetto G, Barthlen W, Defachelles AS, Ferrari A, Weldon CB, Brecht IB, Schneider DT, Bisogno G, Kolenova A, Ben-Ami T, Martinova K, Virgone C, Stachowicz-Stencel T, Kachanov D, Reguerre Y: Pancreatoblastoma in children: EXPeRT/PARTNER diagnostic and therapeutic recommendations. Pediatric blood & cancer 2021, 68 Suppl 4:e29112 [PMID: 34174157]
- Brecht IB, Bremensdorfer C, Schneider DT, Frühwald MC, Offenmüller S, Mertens R, Vorwerk P, Koscielniak E, Bielack SS, Benesch M, Hero B, Graf N, von Schweinitz D, Kaatsch P: Rare malignant pediatric tumors registered in the German Childhood Cancer Registry 2001-2010. Pediatric blood & cancer 2014, 61: 1202 [PMID: 24585499]
- Bisogno G, Ferrari A, Bien E, Brecht IB, Brennan B, Cecchetto G, Godzinski J, Orbach D, Reguerre Y, Stachowicz-Stencel T, Schneider DT: Rare Cancers in Children - The EXPeRT Initiative: A Report from the European Cooperative Study Group on Pediatric Rare Tumors. Klin Pädiatr 2012, 224: 416 [PMID: 23143769]
- Brecht IB, Graf N, Schweinitz D, Frühwald MC, Bielack SS, Schneider DT: Networking for children and adolescents with very rare tumors: foundation of the GPOH Pediatric Rare Tumor Group. Klinische Padiatrie 2009, 221: 181 [PMID: 19437371]