Wilms-Tumor (Nephroblastom) – Kurzinformation
Der Wilms-Tumor (Nephroblastom) ist ein bösartiger Tumor der Niere. In diesem Text erhalten Sie die wichtigsten Informationen zu Krankheitsbild, Häufigkeit, möglichen Ursachen, Symptomen, Diagnose, Therapieplanung, Behandlung sowie zur Prognose der Erkrankung.
Autor: Maria Yiallouros, Redaktion: Maria Yiallouros, Freigabe: Prof. Dr. med. Norbert Graf, Zuletzt geändert: 14.03.2024 https://dx.doi.org/10.1591/poh.wilms-patinfo.kurz.1.20120611
Inhaltsverzeichnis
Krankheitsbild
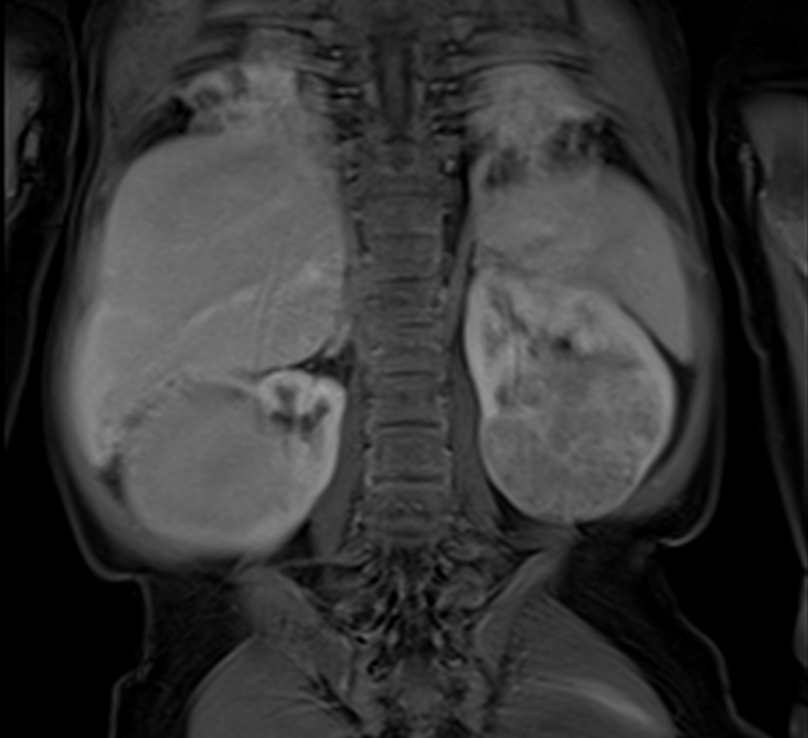
Der Wilms-Tumor, auch Nephroblastom genannt (Nephro- für Niere, -blastom für Tumor), ist ein bösartiger solider Tumor der Niere. Er ist nach dem Heidelberger Chirurgen Max Wilms benannt, der sich um 1900 besonders mit dieser Krankheit befasste und sie ausführlich beschrieb.
Der Wilms-Tumor entsteht durch eine Entartung von primitivem Gewebe. Er erinnert an embryonales Nierengewebe und ist aus unterschiedlichen Gewebearten zusammengesetzt. Meist enthält der Tumor unreife Vorläuferzellen (Blastem) der Niere; es können aber auch andere, unterschiedlich ausgereifte Gewebetypen vertreten sein, so zum Beispiel Bindegewebe, Muskel-, Knorpel- und Epithelgewebe. Wilms-Tumoren werden daher auch als „Mischtumoren“ bezeichnet.
Die Bösartigkeit der Wilms-Tumoren besteht in ihrem raschen Wachstum und in ihrer Neigung, frühzeitig Tochtergeschwülste (Metastasen) zu bilden. Etwa 15 % (in Deutschland dank der Vorsorgeuntersuchungen im Kindesalter nur circa 11 %) der Patienten mit Wilms-Tumor haben bereits bei der Diagnosestellung Metastasen. Hiervon sind vor allem Lymphknoten in der Umgebung der Niere sowie die Lunge betroffen. Nur vereinzelt finden sich Metastasen in der Leber, bei sehr fortgeschrittener Erkrankung auch in den Knochen, dem Zentralnervensystem oder außerhalb des Bauchraumes. Bei ungefähr 5 % der Kinder treten Wilms-Tumoren von Anfang an nicht nur in einer, sondern in beiden Nieren auf. Ausgangsgewebe können in diesen Fällen so genannte nephrogene Reste sein. Darunter versteht man unreifes, embryonales Nierengewebe, das als Vorstufe des Wilms-Tumors gilt.
Häufiger als andere Tumorerkrankungen im Kindesalter tritt das Nephroblastom gemeinsam mit bestimmten weiteren Fehlbildungen und/oder mit erblichen Krebssyndromen auf (siehe Abschnitt „Ursachen“).
Häufigkeit
Der Wilms-Tumor macht, je nach berücksichtigter Altersspanne (0-14 Jahre oder 0-17 Jahre), etwa 5,4 % beziehungsweise 4,2 % aller Krebserkrankungen im Kindes- und Jugendalter aus. Er ist der häufigste Nierentumor bei Kindern und gehört auch insgesamt zu den häufigeren soliden Tumoren dieser Altersgruppe. In Deutschland erkranken nach Angaben des Deutschen Kinderkrebsregisters (Mainz) jährlich etwa 95 Kinder und Jugendliche unter 18 Jahren neu an einem Wilms-Tumor. Damit sind pro Jahr acht bis neun von 1.000.000 Kindern bis zum vollendeten 17. Lebensjahr von dieser Krankheit betroffen.
Da Wilms-Tumoren embryonale Tumoren sind, kommen sie vor allem im frühen Kindesalter vor: Etwa 60 % der Patienten sind zwischen 1 und 4 Jahre alt; 15 % der betroffenen Patienten sind Säuglinge. Das durchschnittliche Erkrankungsalter liegt bei 3 Jahren. Mädchen erkranken etwas häufiger als Jungen. Ein Wilms-Tumor kann aber auch bei älteren Kindern und Jugendlichen vorkommen. Selten tritt er bei Erwachsenen auf.
Ursachen
Die Ursachen für die Entstehung eines Wilms-Tumors sind noch weitgehend ungeklärt. Bekannt ist jedoch, dass an der Entwicklung dieser Tumoren Veränderungen bestimmter Gene und Chromosomen beteiligt sind. Am besten untersucht ist bislang das so genannte Wilms-Tumorgen 1 (WT1-Gen) auf Chromosom 11. Es spielt unter anderem eine Schlüsselrolle bei der normalen Nierenentwicklung und kann, wenn es in veränderter Form vorliegt, zur Tumorbildung und anderen Fehlbildungen führen. Weitere Wilms-Tumorgene sind sowohl auf Chromosom 11 als auch auf anderen Chromosomen gefunden worden. Auch andere chromosomale Veränderungen können das Risiko für die Entwicklung eines Wilms-Tumors erhöhen. Nach heutigem Wissen müssen mehrere genetische Veränderungen (Mutationen) zusammenwirken, bevor ein Wilms-Tumor entsteht.
Wilms-Tumoren kommen häufig (mit 10-15 %) bei Kindern mit einem so genannten Krebsprädispositionssyndrom vor, einer genetisch bedingten Erkrankung, die unter anderem mit einer erblichen Veranlagung für Tumoren einhergeht. Krebsprädispositionssyndrome, die bei der Entstehung eines Wilms-Tumors eine Rolle spielen können, sind zum Beispiel das WAGR-Syndrom, das Beckwith-Wiedemann-Syndrom (BWS) und die Hemihypertrophie, das Denys-Drash-Syndrom (DDS) das Perlman-Syndrom und andere, noch seltenere Syndrome. All diese Syndrome, die insgesamt nur bei einem kleinen Teil der Patienten eine Rolle spielen, sind durch ein erhöhtes Risiko für einen Wilms-Tumor und verschiedene begleitende Fehlbildungen gekennzeichnet.
Darüber hinaus gibt es Familien, in denen Wilms-Tumoren aufgrund unterschiedlicher erblicher Veranlagungen gehäuft auftreten, ohne dass gleichzeitig Anomalien im Rahmen eines Syndroms vorliegen. Das betrifft etwa 1-2 % aller Kinder mit Wilms-Tumor, vornehmlich jene, die an beidseitigen (bilateralen) Tumoren erkrankt sind. Bei der Mehrheit der Patienten tritt die Krankheit allerdings neu (sporadisch) auf. Das heißt, es liegt weder ein erbliches Krebssyndrom noch eine Veranlagung für diese Erkrankung innerhalb der Familie. Umweltfaktoren spielen nach heutigem Erkenntnisstand keine Rolle bei der Entstehung eines Wilms-Tumors.
Gut zu wissen: Für Patienten mit genetisch bedingter Veranlagung für die Entwicklung eines Wilms-Tumors gelten spezielle Vorsorge-Maßnahmen zur frühzeitigen Erkennung eventueller Tumoren. Auch ist eine genetische Beratung bei einem Humangenetiker notwendig (siehe Abschnitt „Diagnose“).
Krankheitszeichen
Wilms-Tumoren bereiten zunächst keine Beschwerden oder Schmerzen. Die betroffenen Kinder haben meist einen vorgewölbten „dicken“ Bauch, der oft zunächst als Zeichen guter Ernährung verkannt wird. In circa 10 % der Fälle tastet der Kinderarzt bei einer Vorsorgeuntersuchung ganz zufällig einen Bauchtumor, ohne dass sonstige Krankheitszeichen (Symptome) vorliegen. Selten sind Bauchschmerz oder blutiger Urin (Hämaturie) das erste Symptom der Erkrankung. In einigen Fällen können neu aufgetretene Rückenschmerzen auf einen Nierentumor hinweisen. Auch Fieber, Verdauungsstörungen (wie Verstopfung oder Durchfall), Gewichtsverlust, Bluthochdruck sowie Husten infolge von Lungen-Metastasen können auftreten. Darüber hinaus ist auf bestimmte begleitende Fehlbildungen oder erbliche Syndrome zu achten (siehe Abschnitt "Ursachen").
Diagnose
Findet der (Kinder-)Arzt durch Krankheitsgeschichte (Anamnese) und körperliche Untersuchung Hinweise auf einen Wilms-Tumor beziehungsweise allgemein auf einen Nierentumor, wird er den Patienten in ein Krankenhaus überweisen, das auf diese Form der Krebserkrankung spezialisiert ist (kinderonkologische Behandlungseinrichtung). Denn bei Verdacht auf einen Nierentumor sind verschiedene Untersuchungen notwendig, zunächst um die Diagnose zu sichern, dann aber auch um festzustellen, um welche Form der Erkrankung es sich genau handelt und wie weit sie sich ausgebreitet hat. Die Klärung dieser Fragen ist Voraussetzung für eine optimale Behandlung und für die Einschätzung der Prognose des Patienten.
Bildgebende Untersuchungen zum Tumornachweis
Die wichtigste Rolle bei der Diagnosestellung spielen – neben der körperlichen Untersuchung – bildgebende Verfahren wie die Ultraschalluntersuchung (Sonographie), die Magnetresonanztomographie (MRT) und gegebenenfalls die Computertomographie (CT) (mit und ohne Kontrastmittel). Mit Hilfe dieser Untersuchungen kann ein „Wilms-Tumor“ mit über 95-prozentiger Sicherheit von anderen möglichen Erkrankungen der Niere unterschieden werden. Hierzu zählen zum einen niereneigene Tumoren wie das Klarzellensarkom, der Rhabdoidtumor der Niere, das Nierenzellkarzinom, das kongenitale mesoblastische Nephrom und weitere sehr seltene Nierentumoren; daneben treten selten andere bösartige Tumoren in der Niere auf, zum Beispiel Lymphome, ein Neuroblastom und, sehr selten, Sarkome. Auch die Größe und Ausbreitung des Tumors, also das Krankheitsstadium, lassen sich sehr genau feststellen.
Voraussetzung für eine möglichst exakte Diagnose ist allerdings, dass die Untersuchungen von guter Qualität sind und von einem erfahrenen Arzt durchgeführt werden. Dies ist deshalb so wichtig, weil in Europa (im Gegensatz zu Nordamerika) die feingewebliche Sicherung der Diagnose, also die Entnahme und Untersuchung von Tumorgewebe, meist erst im Anschluss an eine medikamentöse Vorbehandlung (präoperative Chemotherapie) erfolgt (siehe Abschnitt zur Gewebeentnahme).
Weitere Untersuchungen zur Diagnosesicherung und zur Feststellung von Metastasen
Bisweilen reichen die oben genannten bildgebenden Verfahren allein nicht aus, um einen Wilms-Tumor ausreichend sicher von anderen Erkrankungen, zum Beispiel einem Neuroblastom, abzugrenzen. In diesem Fall können weitere Untersuchungen erforderlich sein: zur Unterscheidung von einem Neuroblastom beispielsweise eine Szintigraphie mit der schwach radioaktiv markierten Substanz 123Iod-meta-Iodbenzylguanidin (123I-MIBG-Szintigraphie) oder auch die Suche nach bestimmten Tumormarkern, die bei Neuroblastomen vorkommen, bei einem Wilms-Tumor aber nicht.
Andere Untersuchungen dienen dem Nachweis beziehungsweise Ausschluss von Metastasen. Für die Suche nach Lungenmetastasen ist eine Computertomographie des Brustraumes notwendig. Bei Verdacht auf Metastasen in Leber, Bauchraum oder Gehirn wird eine Magnetresonanztomographie durchgeführt.
Behandlungsvorbereitende Untersuchungen
Je nach Art der geplanten Behandlung kommen vor Therapiebeginn weitere Untersuchungen hinzu, um Zustand und Funktion bestimmter Organe zu überprüfen. Vor einer Chemotherapie gehört dazu vor allem die Überprüfung der Herzfunktion (Echokardiographie), der Hörfunktion (Audiometrie) und der Nierenfunktion (nuklearmedizinische Nierenfunktionsdiagnostik). Veränderungen, die möglicherweise im Laufe der Therapie auftreten, können aufgrund solcher Ausgangsbefunde besser beurteilt und bei der Behandlung entsprechend berücksichtigt werden.
Gewebeentnahme
Eine Gewebeentnahme (Biopsie) mit anschließender feingeweblicher (histologischer) und molekulargenetischer Untersuchung des Tumorgewebes ist – bei eindeutiger bildgebender Diagnose – erst nach einer vier- bis sechswöchigen präoperativen Chemotherapie erforderlich. Sie erfolgt dann im Rahmen der Operation, bei der der Tumor entfernt wird. Nur in Ausnahmefällen wird bereits zu Beginn Gewebe durch eine Stanz- oder Feinnadelbiopsie entnommen.
Gut zu wissen: Nicht alle der genannten Untersuchungen sind bei jedem Patienten notwendig. Andererseits können eventuell aber auch Untersuchungen hinzukommen, die hier nicht erwähnt wurden. Fragen Sie Ihren behandelnden Arzt oder das Behandlungsteam, welche Untersuchungen bei Ihrem Kind geplant sind und warum die jeweilige Untersuchung erforderlich ist.
Vorsorge bei erblich bedingtem Tumorrisiko
Patienten, die an bestimmten erblichen Veranlagungen für eine Tumorerkrankung (Krebsprädispositionssyndrom) leiden (siehe Abschnitt „Ursachen“), können einen Wilms-Tumor und/oder eine Nephroblastomatose, eine Vorstufe des Wilms-Tumors, entwickeln. Auch das Risiko für einen beidseitigen Tumor (bilaterales Nephroblastom) ist in diesem Fall erhöht. Darüber hinaus kann es bei bestimmten Syndromen (zum Beispiel Denys-Drash-Syndrom) frühzeitig zu einer Nierenfunktionsstörung oder gar einem Nierenversagen kommen.
Aus den genannten Gründen wird für Patienten mit einem prädisponierenden Syndrom oder einer Fehlbildung der Nieren, die mit einem erhöhten Risiko für einen Wilms-Tumor einhergehen, eine vierteljährliche klinische und bildgebende Untersuchung mittels Ultraschall empfohlen. Wegen des erhöhten Risikos einer beidseitigen Tumorentwicklung bei solchen Syndromen werden einseitige Nephroblastome wie beidseitige Nephroblastome behandelt. Für alle Patienten mit einem Syndrom oder einem familiären Nephroblastom empfiehlt sich nach humangenetischer Beratung eine molekulargenetische Abklärung.
Therapieplanung
Nachdem die Diagnose feststeht, erfolgt die Therapieplanung. Um eine möglichst individuelle, auf den Patienten zugeschnittene (risikoadaptierte) Behandlung durchführen zu können, berücksichtigt das Behandlungsteam bei der Planung bestimmte Faktoren, die die Prognose des Patienten beeinflussen (so genannte Risiko- oder Prognosefaktoren).
Die wichtigsten Prognosefaktoren bei Patienten mit einem Wilms-Tumor sind:
- das Krankheitsstadium: das heißt, die Ausbreitung des Tumors zum Zeitpunkt der Diagnose und das Ausmaß der Tumorentfernung im Rahmen der Operation;
- die Unterform des Wilms-Tumors: das heißt, die feingeweblichen Eigenschaften des Tumors, die Aufschluss über sein Wachstumsverhalten und somit seine Bösartigkeit geben können.
Für die Prognose des Patienten ist auch wichtig, wie gut die Erkrankung auf die präoperative Chemotherapie anspricht; Krankheitsstadium und Unterformen des Tumors spielen dabei eine entscheidende, aber nicht die alleinige Rolle. Die Wissenschaftler haben zum Beispiel herausgefunden, dass neben den feingeweblichen auch die molekulargenetischen Eigenschaften des Tumors die Entwicklung der Krankheit beeinflussen können. So scheinen bestimmte Gen- und Chromosomenveränderungen im Tumorgewebe mit einer schlechteren Prognose einherzugehen.
Die verschiedenen Faktoren fließen mit unterschiedlicher Gewichtung in die Behandlungsplanung ein mit dem Ziel, für jeden Patienten durch die Auswahl der jeweils adäquaten Therapie das bestmögliche Behandlungsergebnis zu erreichen.
Einteilung des Wilms-Tumors nach Krankheitsstadien
Das Krankheitsstadium eines Patienten mit Wilms-Tumor ist ein wichtiges Kriterium bei der Wahl der geeigneten Behandlungsstrategie. Bei der Einteilung nach Krankheitsstadien wird berücksichtigt, ob der Tumor die Tumorkapsel schon überschritten hat oder nicht, ob Blutgefäße oder benachbarte Lymphknoten befallen sind oder gar Fern-Metastasen vorliegen und letztlich, ob eine oder beide Nieren betroffen sind.
Wichtig bei der Definition des Stadiums ist außerdem, ob der Tumor durch eine Operation vollständig entfernt werden kann oder nicht. Aus diesem Grund ist die exakte Beurteilung des Krankheitsstadiums erst nach dem operativen Eingriff möglich. Vor Beginn der Behandlung erfolgt (anhand der bildgebenden Untersuchungen zum Zeitpunkt der Diagnose) in einer vorläufigen Einteilung die Unterscheidung zwischen lokalisierten Tumoren in einer Niere, Tumoren mit Metastasen und Tumoren mit beidseitigem Nierenbefall.
Nach der in Deutschland üblichen Stadieneinteilung nach SIOP (der Internationalen Fachgesellschaft für Krebserkrankungen im Kindes- und Jugendalter) werden fünf verschiedene Krankheitsstadien beim Wilms-Tumor unterschieden (siehe Tabelle im Anschluss).
|
Krankheitsstadien |
Definition |
|---|---|
|
Stadium I |
Der Tumor ist auf die Niere beschränkt. Die Tumorkapsel wird nicht überschritten. Der Tumor kann vollständig entfernt werden. |
|
Stadium II |
Der Tumor überschreitet die Tumorkapsel. Der Tumor kann vollständig entfernt werden. Lymphknoten sind nicht befallen. |
|
Stadium III |
Der Tumor kann nicht vollständig entfernt werden oder regionale Lymphknoten sind befallen oder es liegt eine Tumorruptur (Riss in der Gewebestruktur) vor. Es liegen keine Fernmetastasen vor. |
|
Stadium IV |
Es liegen Fernmetastasen vor, unter anderem in Lunge, Leber, Knochen und/oder Gehirn. |
|
Stadium V |
Beidseitiges (bilaterales) Nephroblastom |
Anmerkung zur Stadieneinteilung: Auch für die Stadien IV und V wird ein lokales Stadium (I-III) bestimmt, denn dieses ist ausschlaggebend dafür, welche Therapie nach der Operation (postoperativ) gewählt wird. Wenn ein beidseitiger Wilms-Tumor vorliegt, ist der Tumor mit dem höchsten lokalen Stadium für die Wahl der postoperativen Therapie relevant.
Einteilung des Wilms-Tumors nach feingeweblichen Eigenschaften
Wilms-Tumoren können feingeweblich (histologisch) sehr unterschiedlich aufgebaut sein, je nachdem, aus welchen Gewebearten sie bestehen und wie ausgereift (differenziert) die Zellen der einzelnen Gewebe sind. Der Gewebe-Aufbau des Tumors steht wiederum in engem Zusammenhang damit, wie gut die Erkrankung behandelt werden kann und wie gut folglich die Prognose des Patienten ist. Aus diesem Grund ist es für die Therapieplanung entscheidend zu wissen, welche Unterform (Subtyp) des Wilms-Tumors beim einzelnen Patienten vorliegt – beziehungsweise ob es sich überhaupt um einen Wilms-Tumor oder einen anderen Tumor handelt.
Anhand der feingeweblichen Eigenschaften (Histologie) werden Wilms-Tumoren in drei große Gruppen eingeteilt, die die Bösartigkeit (Malignität) der Krankheit widerspiegeln:
- Nephroblastom – niedriger Malignitätsgrad (günstige Histologie )
- Nephroblastom – intermediärer Malignitätsgrad (Standardhistologie)
- Nephroblastom – hoher Malignitätsgrad (ungünstige Histologie)
Jeder dieser drei Gruppen werden bestimmte Unterformen des Wilms-Tumors zugeordnet. Welche Unterformen das im Einzelnen sind, also wie die Einteilung (Klassifikation) genau erfolgt, richtet sich danach, ob das Tumorgewebe vor oder nach einer Chemotherapie untersucht und bewertet wird. In Deutschland und in anderen europäischen Ländern ist bei der Mehrheit der Patienten Letzteres der Fall: Die Gewebeentnahme und -untersuchung erfolgen nach einer präoperativen Chemotherapie.
Behandlung
Die Behandlung eines Patienten mit Wilms-Tumor muss in einer kinderonkologischen Behandlungseinrichtung erfolgen. Dort ist das hoch qualifizierte Fachpersonal (Ärzte, Fachpflegekräfte) auf die Behandlung krebskranker Kinder spezialisiert und mit den modernsten Therapieverfahren vertraut. Darüber hinaus stehen die Ärzte dieser Klinikabteilungen in fachorientierten Arbeitsgruppen in ständiger, enger Verbindung miteinander und behandeln ihre Patienten nach gemeinsam entwickelten und stetig weiter verbesserten Therapieplänen.
Das Ziel der Behandlung ist, eine Heilung des Patienten zu erreichen und dabei das Risiko therapiebegleitender Nebenwirkungen und Spätfolgen so gering wie möglich zu halten.
Behandlungsmethoden
Die Behandlung eines Patienten mit Wilms-Tumor besteht in erster Linie aus einer Kombination von Operation und Chemotherapie; selten erfolgt zusätzlich eine Strahlentherapie.
Meist wird die Therapie mit einer Chemotherapie eingeleitet, um den Tumor zu verkleinern und somit besser operierbar zu machen. Bei manchen Patienten beginnt die Behandlung mit der Operation. Der chirurgische Eingriff hat zum Ziel, den Tumor und eventuell auch Metastasen zu entfernen. In der Regel schließt sich an die Operation eine (weitere) Chemotherapie an. In Abhängigkeit vom Tumorstadium nach der Operation und/oder bei Metastasen kann zusätzlich eine Bestrahlung der Tumorregion erforderlich sein.
Welche Behandlungsstrategie im Einzelfall angewandt wird, richtet sich nach den Ergebnissen der feingeweblichen Untersuchung (Unterform des Wilms-Tumors) und dem Ausbreitungsstadium des Tumors nach der Operation (Tumorstadium). Je bösartiger der Tumor ist und je weiter fortgeschritten die Krankheit, umso komplexer und intensiver wird die Therapie sein. Da die Behandlung eines Wilms-Tumors mit akuten Nebenwirkungen einhergehen kann, erfolgen während der Behandlung unterstützende Therapiemaßnahmen (Supportivtherapie), die der Vorbeugung und/oder Behandlung dieser Begleiteffekte dienen. Hier finden Sie Informationen zur Supportivtherapie sowie hilfreiche Empfehlungen für zu Hause.
Behandlungsablauf
Die folgenden Therapiephasen werden nach den derzeitigen Behandlungsrichtlinien der internationalen und deutschen Fachgesellschaft für Krebserkrankungen (SIOP und GPOH) unterschieden:
Chemotherapie vor der Operation
In Deutschland und anderen europäischen und außereuropäischen Ländern wird die Therapie bei allen Patienten, die über sechs Monate und unter 16 Jahre alt sind, mit einer Chemotherapie eingeleitet. Es hat sich gezeigt, dass durch eine solche präoperative Chemotherapie der Tumor meist verkleinert und anschließend besser operiert werden kann. Darüber hinaus verringert sich das Risiko, dass der Tumor während der Operation platzt und Tumormaterial im Bauchraum verstreut wird. Auch auf eine Bestrahlung im Anschluss an die Operation kann häufiger verzichtet werden.
Um möglichst alle bösartigen Tumorzellen zu vernichten, wird eine Kombination verschiedener zellwachstumshemmender Medikamente (Zytostatika) eingesetzt, die sich bei der Bekämpfung von Wilms-Tumoren als besonders wirkungsvoll erwiesen haben. Hierzu gehören in erster Linie die Medikamente Vincristin und Actinomycin D (Dactinomycin). Bei manchen Patienten (zum Beispiel mit metastasiertem oder hochmalignem Wilms-Tumor) kommt zusätzlich noch ein Anthrazyklin (Doxorubicin) zum Einsatz. Die Dauer der präoperativen Chemotherapie beträgt in der Regel vier Wochen, bei Patienten mit metastasiertem Wilms-Tumor sechs Wochen und bei beidseitigem Tumor bis zu maximal zwölf Wochen.
Bei Patienten, die an einem beidseitigen Wilms-Tumor erkrankt sind, wird die Behandlungsdauer individuell festgelegt, denn das Ziel ist in diesem Fall vor allem, dass der Tumor beidseits nierenerhaltend operiert werden kann. Zu diesem Zweck kann auch eine Intensivierung der präoperativen Chemotherapie durch weitere Zytostatika (wie Etoposid und Carboplatin) angezeigt sein. Das Gleiche gilt auch für Patienten mit einer Veranlagung für die Entwicklung eines Nephroblastoms (prädisponierendes Syndrom): Auch hier wird der Erhalt von möglichst viel funktionierendem Nierengewebe angestrebt.
Bei Säuglingen unter sechs Monaten und Jugendlichen über 16 Jahre erfolgt vor der Operation keine Chemotherapie. Der Grund dafür ist, dass in diesen Altersgruppen häufig andere Arten von Nierentumoren auftreten (zum Beispiel ein kongenitales (angeborenes) mesoblastisches Nephrom oder ein Nierenzellkarzinom), die anders behandelt werden müssen. Die Operation mit Entfernung des gesamten Tumors und die anschließende Untersuchung durch den Pathologen sind entscheidend, damit die Art des Tumors und das lokale Tumorstadium festgestellt werden können, um dann die richtige Behandlung einzuleiten.
Operation
Mit der Operation wird das Ziel verfolgt, den Tumor vollständig zu entfernen, Tumormaterial für die feingewebliche und molekulargenetische Untersuchung zu gewinnen und gleichzeitig festzustellen, wie weit sich der Tumor ausgebreitet hat.
Die Art der Operation hängt in erster Linie davon ab, ob eine oder beide Nieren betroffen sind.
Liegt ein einseitiger (unilateraler) Wilms-Tumor vor, so wird bei der Operation der Tumor meist mitsamt der Niere, aus der er entstanden ist, entfernt. Dieses Verfahren wird als Tumornephrektomie bezeichnet. Die verbliebene Niere vergrößert sich in den darauffolgenden Wochen und Monaten, so dass sie schließlich auch die Funktion der verlorenen Niere übernehmen kann. Allerdings ist es in diesem Fall wichtig, dass diese Niere im Laufe des Lebens nicht ebenfalls geschädigt wird, beispielsweise durch eine chronische Entzündung. Eine nierenerhaltende Operation kann in manchen Fällen dann erfolgen, wenn der Tumor durch einen erfahrenen Chirurgen komplett aus der Niere entfernt werden kann. Meist liegt dann ein Krankheitsstadium I vor (siehe Abschnitt „Therapieplanung“).
Wenn bereits von Anfang an beide Nieren betroffen sind (bilateraler Wilms-Tumor) oder das Risiko für eine beidseitige Erkrankung hoch ist (zum Beispiel bei einem erblichen Krebsprädispositionssyndrom), müssen die behandelnden Ärzte individuell entscheiden, welche Vorgehensweise für den Patienten die jeweils beste ist, damit zumindest eine der beiden Nieren erhalten werden kann.
Sind nach der präoperativen Chemotherapie noch Lungenmetastasen vorhanden, so können auch diese oft durch einen chirurgischen Eingriff entfernt werden. Wichtig ist, dass jede Operation von einem erfahrenen Kinderchirurgen oder Kinderurologen durchgeführt wird.
Chemotherapie nach der Operation
Nach der Operation wird die Chemotherapie in der Regel fortgesetzt (postoperative Chemotherapie). Lediglich bei Patienten mit einem niedriggradig bösartigen Wilms-Tumor, der ausschließlich auf die Niere begrenzt war und bei der Operation vollständig entfernt werden konnte (Tumorstadium I), ist die Behandlung nach der Operation abgeschlossen. Alle anderen Patienten erhalten – je nach Tumortyp, Tumorgewicht zum Zeitpunkt der Operation und lokalem Tumorstadium – eine mehr oder weniger intensive und lang andauernde Chemotherapie:
Bei intermediär bösartigen Tumoren ohne Fernmetastasen (Tumorstadien I bis III) wird die Behandlung meist mit nur zwei Zytostatika (Vincristin und Actinomycin-D) fortgeführt. Bei sehr bösartiger Krankheit und höheren Tumorstadien kommen bis zu vier Zytostatika zum Einsatz (zum Beispiel eine Kombination aus Doxorubicin, Carboplatin, Etoposid und Cyclophosphamid). Die Therapiedauer nach Operation beträgt zwischen einem Monat (bei Patienten mit Wilms-Tumor intermediärer Bösartigkeit und Tumorstadium I) und acht bis zehn Monaten (bei Patienten mit sehr bösartigen Wilms-Tumor-Formen oder Metastasenresten nach der Operation). Bei beidseitigen Nierentumoren kann die Behandlung bis zu einem Jahr dauern, wenn nephrogene Reste (Vorstufen des Wilmstumors) vom Pathologen nachgewiesen wurden.
Strahlentherapie
Durch die Entwicklung wirksamer Chemotherapiekombinationen und die Einführung der präoperativen Chemotherapie kann inzwischen bei den meisten Patienten auf eine Strahlentherapie verzichtet werden. Manche Patienten benötigen allerdings nach wie vor eine Bestrahlung im Anschluss an Operation und Chemotherapie. Dazu zählen Patienten mit einem intermediär bösartigen Wilms-Tumor im Stadium III und Patienten mit hochgradig bösartigen Wilms-Tumoren ab Stadium II (siehe Abschnitt „Therapieplanung“).
Die Bestrahlung erfolgt durch die Haut auf die Tumorregion mit einer Strahlendosis von 15 bis 30 Gray (Gy), in Abhängigkeit von der feingeweblichen (histologischen) Untersuchung nach der Operation. Wenn noch Tumorreste vorhanden sind, wird die Strahlendosis auf das verbliebene Tumorareal erhöht. Bei einem Platzen (Ruptur) des Tumors während der Operation muss der gesamte Bauchraum, wiederum in Abhängigkeit von der Histologie, mit 20 oder 30 Gy bestrahlt werden. Patienten, bei denen nach der präoperativen Chemotherapie und der Operation noch Lungenmetastasen nachweisbar sind, erhalten auch eine Bestrahlung des Brustraumes. Bei beidseitigen (bilateralen) Tumoren und vorhandener Restniere kann die Strahlendosis bei intermediärer Bösartigkeit auch herabgesetzt sein (unter 12 Gy), um die noch bestehende Nierenfunktion möglichst langfristig zu erhalten.
Therapieoptimierungsstudien / Register
In den großen Behandlungszentren werden Kinder und Jugendliche mit einem Wilms-Tumor nach standardisierten Therapieprotokollen behandelt. Sie alle haben zum Ziel, die Langzeitüberlebensraten der Patienten zu verbessern und gleichzeitig therapiebedingte Spätfolgen so gering wie möglich zu halten. Die Behandlung nach solchen Therapieprotokollen erfolgt in aller Regel im Rahmen von Therapieoptimierungsstudien.
Im deutschsprachigen Raum ist im August 2011 eine langjährige Therapieoptimierungsstudie abgelaufen, an der sich ungefähr 100 Kinderkliniken in ganz Deutschland, Österreich und der Schweiz beteiligt hatten: die Studie SIOP WT 2001/GPOH. Die Studie wurde von der Nephroblastom-Studienzentrale der deutschen Fachgesellschaft für Krebserkrankungen im Kindes- und Jugendalter – der Gesellschaft für Pädiatrische Onkologie und Hämatologie, GPOH – durchgeführt und war Teil der Internationalen Nephroblastomstudie der SIOP (International Society of Pediatric Oncology).
Inzwischen ist, auf Grundlage der Ergebnisse bisheriger nationaler und internationaler Studien, eine neue SIOP-Studie für Kinder und Jugendliche mit Nierentumoren entwickelt worden: die Studie UMBRELLA SIOP-RTGS 2016. Sie ist in einigen europäischen Ländern bereits 2019 und in Deutschland Ende 2021 teilweise angelaufen. Bis die Studie auch in Deutschland in allen Zentren starten kann, werden Patienten mit Wilms-Tumor weiterhin in noch nicht teilnehmenden Zentren in einem Register erfasst und nach Empfehlungen behandelt, die sich im Rahmen der letzten Studie als erfolgreich erwiesen haben. Die deutsche Studien- und Registerzentrale befindet sich an der Universitäts-Kinderklinik in Homburg/Saar (Studienleitung: Prof. Dr. med. Norbert Graf).
Für Patienten mit einer metastasierten Erkrankung gibt es seit Anfang 2022 die randomisierte Studie RANDOMET 2017. Sie untersucht im Rahmen der präoperativen Chemotherapie, ob die Kombination von Vincristin, Carboplatin und Etoposid gleich gut ist wie die Standardtherapie mit Vincristin, Actinomycin und Doxorubicin. Ziel ist es, auch bei Patienten mit einer metastatischen Erkrankung die Nebenwirkungen durch Doxorubicin am Herzen zu reduzieren, ohne dadurch die Heilungsrate zu verringern.
Besonderheit Register: Patienten, die an keiner Studie teilnehmen, entweder weil zum Zeitpunkt ihrer Erkrankung keine Studie verfügbar ist oder weil sie die Einschlusskriterien einer bestehenden Studie nicht erfüllen, werden oft in einem so genannten Register dokumentiert. Diese dienen zunächst dazu, die Therapie der Patienten wissenschaftlich zu begleiten. Zur Sicherung der optimalen Behandlung verfasst darüber hinaus die jeweilige Studiengruppe in der Regel detaillierte Empfehlungen und berät die behandelnden Ärzte bei der Auswahl der optimalen Therapie für den einzelnen Patienten.
Prognose
Die Heilungsaussichten von Kindern und Jugendlichen mit einem Wilms-Tumor sind sehr gut. Dank der heute eingesetzten modernen Untersuchungsmethoden und der standardisierten Kombinationstherapien können insgesamt über 90 % aller Patienten mit dieser Erkrankung langfristig geheilt werden.
Die Prognose für den einzelnen Patienten hängt allerdings in erster Linie davon ab, welche Unterform des Wilms-Tumors vorliegt und wie weit die Krankheit zum Zeitpunkt der Diagnose fortgeschritten ist (Krankheitsstadium). In der Regel sind die Heilungschancen umso besser, je weniger bösartig der Tumor ist und je früher der Tumor entdeckt wird. So haben beispielsweise Patienten mit einem nicht-metastasierten Tumor niedriger oder intermediärer Bösartigkeit Überlebenschancen von über 90 %, während sehr bösartige Tumoren eine deutlich ungünstigere Prognose aufweisen. Auch bei Patienten mit einem beidseitigen (bilateralen) Wilms-Tumor, ungünstigen molekularen Tumoreigenschaften oder einem Krankheitsrückfall liegen die Heilungsraten in der Regel unter 90 %.
Allerdings können Patienten trotz höherer Krankheitsstadien unter bestimmten Bedingungen noch gute Heilungschancen haben. Bei Patienten mit Fern-Metastasen (Krankheitsstadium IV) zum Beispiel hängt die Prognose entscheidend davon ab, wie gut der Tumor auf die Chemotherapie anspricht: Wird durch die präoperative Chemotherapie und die operative Tumorentfernung eine komplette Tumorrückbildung erreicht, werden Heilungsraten von über 80 % erzielt.
Anmerkung: Bei den genannten Überlebensraten handelt es sich um statistische Größen. Sie stellen nur für die Gesamtheit der an einem Wilms-Tumor erkrankten Patienten eine wichtige und zutreffende Aussage dar. Ob der einzelne Patient geheilt werden kann oder nicht, lässt sich aus der Statistik nicht vorhersagen. Wenn Sie Fragen zur prognostischen Einschätzung der Erkrankungsart Ihres Kindes haben, wenden Sie sich daher bitte an Ihr Behandlungsteam.
 PDF-Datei der Patienten-Kurzinformation zum Wilms-Tumor (457KB)
PDF-Datei der Patienten-Kurzinformation zum Wilms-Tumor (457KB)Verwendete Basisliteratur 
- Erdmann F, Kaatsch P, Grabow D, Spix C: German Childhood Cancer Registry - Annual Report 2019 (1980-2018). Institute of Medical Biostatistics, Epidemiology and Informatics (IMBEI) at the University Medical Center of the Johannes Gutenberg University Mainz 2020 [URI: https://www.kinderkrebsregister.de/ typo3temp/ secure_downloads/ 42507/ 0/ 1c5976c2ab8af5b6b388149df7182582a4cd6a39/ Buch_DKKR_Jahresbericht_2019_komplett.pdf]
- Furtwängler R, Graf N: Nierentumoren, in: Niemeyer C, Eggert A (Hrsg.): Pädiatrische Hämatologie und Onkologie. Springer-Verlag GmbH GDeutschland 2006, 2018 2. vollständig überarbeitete Auflage 2018, 441 [ISBN: 978-3-662-43685-1]
- Graf N: Nephroblastom (Wilms-Tumor). S1-Leitlinie 025/004 AWMF online 2016 [URI: https://www.awmf.org/ uploads/ tx_szleitlinien/ 025-004l_S1_Nephroblastom_Wilms-Tumor_2016-06-abgelaufen.pdf]
- Dome JS, Graf N, Geller JI, Fernandez CV, Mullen EA, Spreafico F, Van den Heuvel-Eibrink M, Pritchard-Jones K: Advances in Wilms Tumor Treatment and Biology: Progress Through International Collaboration. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2015 Sep 20; 33: 2999 [PMID: 26304882]
- Szavay P, Fuchs J, Leuschner I, Selle B, Graf N: Nierentumoren, in: Fuchs J (Hrsg.): Solide Tumoren im Kindesalter, Grundlagen – Diagnostik – Therapie. Schauttauer GmbH 2012, 111 [ISBN: 978-3-7945-2786-1]